Using GraphINVENT to generate novel DRD2 actives
I have been writing a lot about how to use SMILES together with deep learning architectures such as RNNs and LSTM networks to perform various cheminformatic and drug discovery tasks such as de novo design and QSAR modelling. SMILES notation seems to work well together with LSTM networks and is very convenient as most cheminformatic tools can convert them to and from molecular objects and other formats. This provides efficient handling of the tasks, datasets, and generated molecules. With data augmentation tricks, we can further improve the performance.
But what if there was a better way to present and generate molecules than via the SMILES format? After all, molecules are not naturally linear as they contain rings and branches and stereocenters. The graph model with atoms as nodes and bonds as edges is probably closer to the real molecules and a more true format. But how can they be vectorized and read into a neural network? And how can the neural networks generate new molecular graphs? Rocío Mercado has worked with this particular field as a postdoc in the group and I have the pleasure of welcoming her as Cheminformania’s very first guest blogger 🙂 Read on below for a practial guide on how to use the open source code she created for generating novel molecules with predicted bioactivity.
Esben Jannik Bjerrum

Using GraphINVENT to generate novel DRD2 actives
Introduction to GraphINVENT

PhD Rocío Mercardo
Recently, we in the Molecular AI (MAI) team at AstraZeneca published GraphINVENT, a platform for graph-based molecular generation using graph neural networks (GNNs). GraphINVENT uses a tiered deep neural network architecture to probabilistically generate new molecules a single bond at a time and runs on a CUDA-enabled GPU.
In this blog post we would like to introduce how GraphINVENT learns to build molecules resembling a training set of molecules, without any explicit programming of chemical rules. Graph-based generative models are highly flexible, promising tools for addressing challenges in molecular design, but have been relatively less explored for molecular generation compared to string-based methods. Graph-based generative models are exciting tools for molecular design as there are many advantages to working directly with the graph representation that string representations do not have. Nonetheless, graph-based methods are indeed more computationally demanding than string-based methods.
In this paper, we show how the best GNN implemented in GraphINVENT, the GGNN, compares well with state-of-the-art generative models. In this post, the goal is to show you how to use the GGNN model to generate new graphs using a dataset made up of ~4000 dopamine receptor D2 (DRD2) known active molecules. Generating DRD2 actives has become a standard benchmark for molecular generative models.
Downloading the code
GraphINVENT is available at https://github.com/MolecularAI/GraphINVENT. To get a local copy, you can either download it directly by clicking the green code button and selecting Download Zip, or, if you have Git installed, you can clone the repo directly by typing into your command line:
git clone https://github.com/MolecularAI/GraphINVENT.git
The DRD2 actives dataset
A dataset of DRD2 actives has been obtained from ExCAPE-DB and preprocessed as in this nice publication. In summary, compounds with a measured pXC50 > 5 were labeled as actives and split into three sets (training, testing, validation); assuming one is in the base GraphINVENT directory, these can be found in data/DRD2_actives/. Inactive molecules were removed from this dataset. Each split thus contains the following number of active molecules:
- train.smi : 3448
- test.smi : 432
- valid.smi : 431
We will use the DRD2 actives to generate new DRD2 actives using GraphINVENT. To do this, we will need to run three separate GraphINVENT jobs:
- a preprocessing job
- a training job
- a generation job
Each of these jobs is decribed in detail below.
Preprocessing the dataset
Before training any models in GraphINVENT on the DRD2 dataset, it must first be preprocessed. To do this, we will need to know the following information:
- maximum number of nodes per molecule in DRD2_actives
- which atom types are present in DRD2_actives
- which formal charges are present in DRD2_actives
We have provided a few scripts to help calculate these properties in /tools/.
# concatenate the structures in all three splits (for analysis) !cat data/DRD2_actives/train.smi data/DRD2_actives/test.smi data/DRD2_actives/valid.smi >> data/DRD2_actives/all.smi # get the maximum number of nodes in the dataset !python tools/max_n_nodes.py --smi data/DRD2_actives/all.smi # get the atom types present in the dataset !python tools/atom_types.py --smi data/DRD2_actives/all.smi # get the formal charges present in the dataset !python tools/formal_charges.py --smi data/DRD2_actives/all.smi # then, remove `all.smi` as it is no longer needed !rm data/DRD2_actives/all.smi
* Max number of atoms in input file: 72
Done.
* Atom types present in input file: ['C', 'N', 'O', 'F', 'S', 'Cl', 'Br']
Done.
* Formal charges present in input file: [-1, 0, 1]
Done.
Once we know these values, we can move on to preparing a submission script. A sample submission script submit.py has been provided. We begin by modifying the submission script to specify where the dataset can be found and what type of job we want to run. For example:
dataset = "DRD2_actives" # dataset name should correspond to `data/{dataset}`
job_type = "preprocess" # "preprocess", "train", or "generate"
jobname = "preprocessing_example"
data_path = "./data/"
Then, details regarding the specific dataset we want to use need to be defined:
params = {
"atom_types": ["C", "N", "O", "F", "S", "Cl", "Br"],
"formal_charge": [-1, 0, +1],
"max_n_nodes": 72,
"job_type": job_type,
"dataset_dir": f"{data_path}{dataset}/",
}
At this point, we are done defining the job parameters and are ready to submit a preprocessing job. We can do this by simply running the cell below. What this cell does is:
- Create a job directory where we will write any input and output for the preprocessing job.
- Write an input.csv file to the directory; this is an input file which specifies non-default parameters and hyperparameters to use for the job. If there is an input.csv in the specified job_dir, then GraphINVENT will use those settings to run the job.
- Run the preprocessing job from the command line.
In the example below, the input file will be written to output_DRD2_actives/preprocessing_example/.
import os
from submit import write_input_csv
# create the output directory
data_path_minus_data = data_path[:-5]
dataset_output_path = f"{data_path_minus_data}output_{dataset}"
dataset_output_path = os.path.join(dataset_output_path, jobname)
print(f"* Creating dataset directory {dataset_output_path}/")
os.makedirs(dataset_output_path)
# specify the output directory in `params`
params["job_dir"] = f"{dataset_output_path}/"
# write the `input.csv` file
write_input_csv(params_dict=params, filename="input.csv")
# submit the job (using the command line)
!python graphinvent/main.py --job-dir output_DRD2_actives/preprocessing_example/
* Creating dataset directory ./output_DRD2_actives/preprocessing_example/
* Run mode: 'preprocess'
* Preprocessing validation data.
-- 431 molecules in set.
-- 14537 total subgraphs in set.
Datasets resized.
* Resaving datasets in unchunked format.
100%|█████████████████████████████████████████████| 3/3 [00:00<00:00, 33.47it/s]
-- time elapsed: 111.76775 s
* Preprocessing test data.
-- 432 molecules in set.
-- 14525 total subgraphs in set.
Datasets resized.
* Resaving datasets in unchunked format.
100%|█████████████████████████████████████████████| 3/3 [00:00<00:00, 26.88it/s]
-- time elapsed: 222.72112 s
* Preprocessing training data.
-- 3448 molecules in set.
-- 116863 total subgraphs in set.
Datasets resized.
Writing training set properties.
Shuffling training dataset.
* Resaving datasets in unchunked format.
100%|█████████████████████████████████████████████| 3/3 [00:00<00:00, 4.63it/s]
-- time elapsed: 1269.28145 s
The preprocessing job usually takes a few minutes to finish (1269 seconds, or 21 minutes, above).
During preprocessing jobs, the following will be written to data/DRD2_actives/:
- 3 HDF files (train.h5, valid.h5, and test.h5)
- preprocessing_params.csv, a file containing parameters used in preprocessing the dataset (for later reference)
- train.csv, a file containing training set properties (e.g. histograms of number of nodes per molecule, number of edges per node, etc)
Indeed, we can check that these files were indeed created by typing:
!ls -lh data/DRD2_actives/
total 2.5G
-rw-rw---- 1 rocio cc 243 Sep 18 08:34 preprocessing_params.csv
-rw-rw---- 1 rocio cc 245M Sep 18 08:38 test.h5
-rw-rw---- 1 rocio cc 21K Sep 17 16:12 test.smi
-rw-rw---- 1 rocio cc 1019 Sep 18 08:55 train.csv
-rw-rw---- 1 rocio cc 2.0G Sep 18 10:26 train.h5
-rw-rw---- 1 rocio cc 168K Sep 17 16:12 train.smi
-rw-rw---- 1 rocio cc 249M Sep 18 10:26 valid.h5
-rw-rw---- 1 rocio cc 21K Sep 17 16:12 valid.smi
You can see that the HDF files created are significantly larger than their corresponding SMILES files, as they contain all the processed molecules in matrix representation, as well as the target properties (the action probability distributions) that will be used during training.
Training on the DRD2 actives
When the DRD2_actives dataset is done preprocessing, we are ready to train models with it.
For this example we will use the GGNN model along with the best parameters and hyperparameters described in the paper (using parameters from the MOSES dataset).
You can modify the same submit.py script to instead run a training job using the dataset. Begin by changing the job_type and jobname; the dataset variable should of course remain the same.
dataset = "DRD2_actives" job_type = "train" jobname = "training_example"
We then need to specify all parameters that we want to use for training. If any parameters are not specified before running a job, GraphINVENT will use the default values specified in graphinvent/parameters/defaults.py.
params = {
"atom_types": ["C", "N", "O", "F", "S", "Cl", "Br"],
"formal_charge": [-1, 0, +1],
"max_n_nodes": 72,
"job_type": job_type,
"dataset_dir": f"{data_path}{dataset}/",
"min_rel_lr": 1e-3,
"model": "GGNN",
"lrdf": 0.9999,
"lrdi": 1000,
"sample_every": 10,
"batch_size": 500,
"init_lr": 1e-4,
"epochs": 100,
"block_size": 50000,
"n_samples": 5000,
}
Note that, if you plan to use different datasets with GraphINVENT, it is not always the case that the “default” values will work well for a given new dataset. For instance, the parameters related to the learning rate decay are strongly dependent on the dataset used, and you might have to tune them to get optimal performance using your datasets. Depending on your system, you might also need to tune the mini-batch and/or block size so as to reduce/increase the memory requirement for training jobs. Details on this are available here and here.
Using the above parameters, we can run a GraphINVENT training job by running the cell below (it took about 1.5 hours on my workstation, which has an NVIDIA GeForce RTX 2080 Ti). What the cell below does is, again:
- Create a job directory where we will write any input and output for the preprocessing job.
- Write an input.csv file to the directory; this is an input file which specifies non-default parameters and hyperparameters to use for the job.
- Run the training job from the command line, piping all output into training-output.
- Print only the last few lines of training-output.
In the example below, the input file will be written to output_DRD2_actives/training_example/. Training output is suppressed (only the last couple lines are shown).
# create the output directory
data_path_minus_data = data_path[:-5]
dataset_output_path = f"{data_path_minus_data}output_{dataset}"
dataset_output_path = os.path.join(dataset_output_path, jobname)
print(f"* Creating dataset directory {dataset_output_path}/")
os.makedirs(dataset_output_path)
# specify the output directory in `params`
params["job_dir"] = f"{dataset_output_path}/"
# write the `input.csv` file
write_input_csv(params_dict=params, filename="input.csv")
# submit the job (using the command line)
!python graphinvent/main.py --job-dir output_DRD2_actives/training_example/ > training-output
# a lot of output is written during training, let's just see the last bit
!tail training-output
* Saving model state at Epoch 100.
-- time elapsed: 5058.75186 s
As the models are training, you will see the progress bar updating on the terminal every epoch. The training status will be saved every epoch to the job directory, output_DRD2_actives/training_example/. Additionally, the evaluation scores will be saved every 10 epochs (sample_every = 10) to the job directory. Among the files written to this directory will be:
- generation.csv, a file containing various evaluation metrics for the generated set, calculated during evaluation epochs
- convergence.csv, a file containing the loss and learning rate for every epoch
- validation.csv, a file containing model scores (e.g. NLLs, UC-JSD) calculated during evaluation epochs
- model_restart_{epoch}.pth, which are the model states for use in restarting jobs, or running generation/validation jobs with a trained model
- generation/, a directory containing structures generated during evaluation epochs (*.smi), as well as information on each structure’s NLL (*.nll) and validity (*.valid)
It is good to check the generation.csv to verify that the generated set features indeed converge to those of the training set (first entry). If they do not, then you will have to tune the hyperparameters to get better performance:
!head -n 1 output_DRD2_actives/training_example/generation.csv | awk '{ print $1 $2 $5 $6 $7}' > quick-check
!grep "Training set," output_DRD2_actives/training_example/generation.csv | awk '{ print $1 $2 $3 $6 $7 $8 }' >> quick-check
!grep "Epoch 100," output_DRD2_actives/training_example/generation.csv | awk '{ print $1 $2 $3 $6 $7 $8 }' >> quick-check
!cat quick-check | column -t -s,
set fraction_valid run_time avg_n_nodes avg_n_edges
Trainingset 1.000 NA 28.868 2.213
Epoch100 0.848 4999.44 27.426 2.207
Indeed, it appears the training set properties (avg_n_nodes and avg_n_edges) match the properties of graphs generated after 100 training epochs. Although it appears that only ~85% of the generated molecules were valid chemical structures, this is not a problem because the action space is huge (generated graphs can have up to 72 heavy atoms). This is an acceptable percent validity value for this model.
It is also good to check the loss to make sure it is smoothly decreasing during training. To do this, we can check the convergence.csv file. If there are any large spikes in the loss, it means the learning rate was not decreased quickly enough. Here we check that the loss has indeed converged (and there were no weird spikes):
!head -n 5 output_DRD2_actives/training_example/convergence.csv > quick-check !echo "..." >> quick-check !tail -n 5 output_DRD2_actives/training_example/convergence.csv >> quick-check !cat quick-check | column -t -s,
epoch lr avg_loss model_score
Epoch 1 0.00010000 3.28330183 NA
Epoch 2 0.00010000 2.05745625 NA
Epoch 3 0.00010000 1.69959128 NA
Epoch 4 0.00010000 1.47861886 NA
...
Epoch 96 0.00000014 0.16531540 NA
Epoch 97 0.00000014 0.16509235 NA
Epoch 98 0.00000014 0.16543394 NA
Epoch 99 0.00000014 0.16489927 NA
Epoch 100 0.00000014 0.16543923 -0.000538
Looks good! Note that the model score only prints every 10 epochs, and hence the model score here only shows up for Epoch 100.
Generating molecules using the trained model
Once we have trained a model, we can use a saved state (e.g. model_restart_100.pth) to generate molecules. To do this, submit.py needs to be updated to specify a generation job. The first setting that needs to be changed is the job_type; all other settings here should be kept fixed so that the program can find the correct job directory:
dataset = "DRD2_actives" job_type = "generate" jobname = "training_example" # keep the same job name as for training, so GraphINVENT can find saved model
We will then need to update the generation_epoch (2nd to last parameter below) to specify the epoch we want to use for generating new structures (100), as well as the n_samples (last parameter below) to specify how many structures we would like to generate (100,000):
params = {
"atom_types": ["C", "N", "O", "F", "S", "Cl", "Br"],
"formal_charge": [-1, 0, +1],
"max_n_nodes": 72,
"job_type": job_type,
"dataset_dir": f"{data_path}{dataset}/",
"min_rel_lr": 1e-3,
"model": "GGNN",
"lrdf": 0.9999,
"lrdi": 1000,
"sample_every": 10,
"batch_size": 500,
"init_lr": 1e-4,
"epochs": 100,
"block_size": 50000,
"generation_epoch": 100,
"n_samples": 100000,
}
All other parameters should be kept the same. Parameters related to training, such as epochs or init_lr, will be ignored during generation jobs.
During generation, molecules will be built in batches of size batch_size. If any memory problems are encountered during generation jobs, reducing the batch size should once again solve them. Generated structures, along with their corresponding metadata, will be written to the generation/ directory within the existing job directory. These files are:
- epochGEN{generation_epoch}_{batch}.smi, containing molecules generated at the epoch specified
- epochGEN{generation_epoch}_{batch}.nll, containing their respective NLLs
- epochGEN{generation_epoch}_{batch}.valid, containing their respective validity (0: invalid, 1: valid)
Additionally, the generation.csv file will be updated with the various evaluation metrics for the generated structures.
We can run a generation job by simply running the cell below, which once again first defines the output directory and then writes an input.csv file specifying the parameters to use before submitting the job.
# define the output directory (already created in previous job)
data_path_minus_data = data_path[:-5]
dataset_output_path = f"{data_path_minus_data}output_{dataset}"
dataset_output_path = os.path.join(dataset_output_path, jobname)
# specify the output directory in `params`
params["job_dir"] = f"{dataset_output_path}/"
# write the `input.csv` file
write_input_csv(params_dict=params, filename="input.csv")
# submit the job (using the command line)
!python graphinvent/main.py --job-dir output_DRD2_actives/training_example/ > generation-output
# a lot of output is written during training, let's just see the last bit
!tail generation-output
-- time elapsed: 988.82848 s
It takes roughly 15 minutes to generate 100,000 molecules resembling the DRD2 actives in GraphINVENT.
Visualizing generated molecules and predicting their activities
Now that we have generated 100,000 molecules, we would like to see what these molecules look like and analyze their activity.
To make things more convenient for any subsequent analyses, you can make an analysis/ directory and concatenate all structures generated in different batches into one file, as follows:
!mkdir output_DRD2_actives/analysis/ !for i in output_DRD2_actives/training_example/generation/epochGEN100_*.smi; do cat $i >> output_DRD2_actives/analysis/epochGEN100.smi; done
Even if the models are well trained, “Xe” and empty graphs may still appear in the generated structures, as there is always a small probability of sampling invalid actions. We don’t necessarily want to include invalid entries in our analysis, so these can be filtered out as follows:
!sed "/Xe/d" output_DRD2_actives/analysis/epochGEN100.smi > output_DRD2_actives/analysis/epochGEN100_validonly.smi # remove "Xe" placeholders from file !sed -i "/^ [0-9]\+$/d" output_DRD2_actives/analysis/epochGEN100_validonly.smi # remove empty graphs from file !sed -i "/SMILES/d" output_DRD2_actives/analysis/epochGEN100_validonly.smi # remove lines that say "SMILES" (headers)
To visualize 24 random molecules from the set of valid molecules generated, we can run the cell below.
import random
import rdkit
from rdkit.Chem.Draw import MolsToGridImage
from rdkit.Chem.rdmolfiles import SmilesMolSupplier
smi_file = "output_DRD2_actives/analysis/epochGEN100_validonly.smi"
# load molecules from file
mols = SmilesMolSupplier(smi_file, sanitize=True, nameColumn=-1)
n_samples = 24
mols_list = [mol for mol in mols]
mols_sampled = random.sample(mols_list, n_samples)
png_filename=smi_file[:-3] + "png" # name of PNG file to create
labels=list(range(n_samples)) # label structures with a number
# draw the molecules (creates a PIL image)
img = MolsToGridImage(mols=mols_sampled,
molsPerRow=6,
legends=[str(i) for i in labels])
img.save(png_filename)
# display
from IPython.display import Image
Image(filename=png_filename)
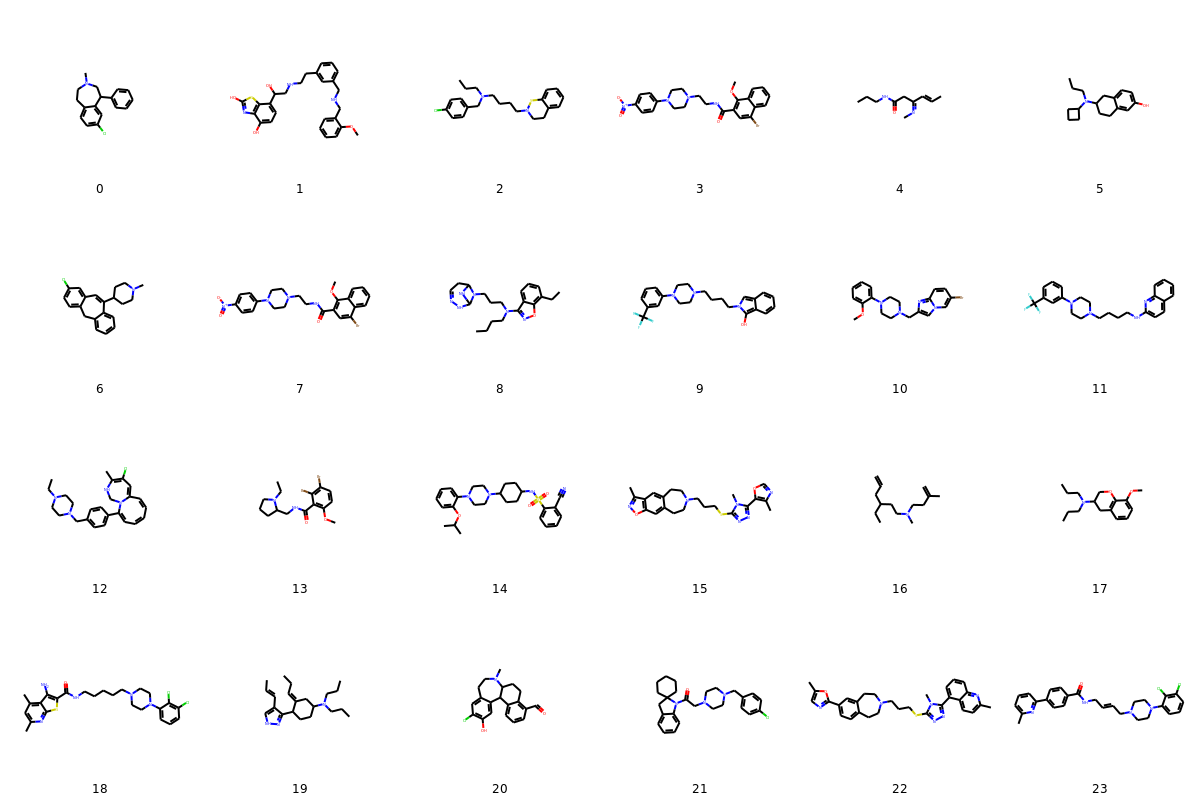
The molecules look pretty good! Some clearly better than others.
As such, let’s analyze how many of these structures are potentially new actives. To do this, we will use the QSAR model available in this repo under models/qsar_model.pickle.
To follow along, you can download the code and follow the instructions at https://github.com/pcko1/Deep-Drug-Coder. Note that you will need to install git-lfs in order to download the models. Besides installing git-lfs, you will also need to configure the virtual environment for running the QSAR model, called ddc_env.
Validity
First, let’s analyze the validity of the molecules generated:
# analyze the validity of the structures
all_smi = "output_DRD2_actives/analysis/epochGEN100.smi"
total_n_samples = len(open(all_smi).readlines())
valid_smi = "output_DRD2_actives/analysis/epochGEN100_validonly.smi"
valid_n_samples = len(open(valid_smi).readlines())
percent_valid = valid_n_samples/total_n_samples * 100
print(f"Total structures sampled: {total_n_samples}")
print(f"Valid structures sampled: {valid_n_samples} ({percent_valid:.1f}%)")
Total structures sampled: 100701
Valid structures sampled: 85197 (84.6%)
As we can see, only about 85% of the generated structures are valid. This is not bad, and in fact is quite great, considering the huge output space which the generative models had to learn: molecules containing up to 72 (!!!) heavy atoms, with 7 different heavy atom types, 3 formal charge states, and 3 bond types represented in this space. Furthermore, remember that no further hyperparameter optimization (HO) was performed on the cGGNN; we simply took the best hyperparameters identified in the paper. As such, the percent validity achieved could perhaps be higher after further HO on this dataset, but for this small experiment we will not worry about that.
Activity
More interestingly, we would like to predict the activity of the structures generated using a pre-trained QSAR model. For details on the QSAR model, please see the original paper. For our purposes, we are considering a predicted activity of > 0.5 (by the model) to be “active”. We will also require molecules to have a Quantitative Estimate of Druglikeness (QED) score > 0.5 in order to be labeled as “active.”
# find which molecules generated were active
import numpy as np
import matplotlib.pyplot as plt
import rdkit
from rdkit import Chem, DataStructs
from rdkit.Chem import AllChem, QED
from rdkit.Chem.rdmolfiles import SmilesMolSupplier, SmilesWriter
import h5py, pickle
# load the QSAR model
with open("../../Deep-Drug-Coder/models/qsar_model.pickle", 'rb') as file:
model_dict = pickle.load(file)
drd2_model = model_dict["classifier_sv"]
# define a function for using the QSAR model to predict activities in molecules
def find_actives(smi_file, activity_threshold=0.5, verbose=True):
""" Predict the activities of molecules in an input SMILES file and output stats.
"""
# load the generated structures
mols_in = SmilesMolSupplier(smi_file, sanitize=True, titleLine=False)
# calculate the properties of the generated structure and compare
active_mols = []
all_activities = []
active_mols_activities = []
for mol in mols_in:
# if a `Mol` object is valid, calculate its activity and QED score
if mol:
qed = QED.qed(mol)
# calculate fingerprint
fp = AllChem.GetMorganFingerprintAsBitVect(mol, 2, nBits=2048)
ecfp4 = np.zeros((2048,))
DataStructs.ConvertToNumpyArray(fp, ecfp4)
# predict activity and pick only the second component
active = drd2_model.predict_proba([ecfp4])[0][1]
all_activities.append(active)
if active > activity_threshold and qed > 0.5:
active_mols.append(mol)
active_mols_activities.append(active)
else:
pass
# print the average activities
average_activity_score = np.mean(all_activities)
if verbose:
print(f"Average activity score: {average_activity_score:.5f}")
# write the active molecules only
actives_smi_file = smi_file[:-4] + "_actives.smi"
writer = SmilesWriter(actives_smi_file)
for mol in active_mols:
writer.write(mol)
if verbose:
# plot a histogram of the activities
_ = plt.hist(all_activities, bins=10)
plt.xlabel("Activity scores")
plt.ylabel("Count")
activities_histogram_file = smi_file[:-4] + "_activities.png"
plt.savefig(activities_histogram_file)
# print a few other stats
percent_active = len(active_mols)/len(mols_in) * 100
total_input_molecules = len(mols_in)
print(f"Total input molecules: {total_input_molecules}")
total_actives = len(active_mols)
print(f"Total actives found (activity score > {activity_threshold}): {total_actives} ({percent_active:.1f}%)")
return None
# predict the activities of molecules in the generated set (valid molecules only)
generated_smiles = "output_DRD2_actives/analysis/epochGEN100_validonly.smi"
find_actives(smi_file=generated_smiles)
Average activity score: 0.76497
Total input molecules: 85197
Total actives found (activity score > 0.5): 48177 (56.5%)
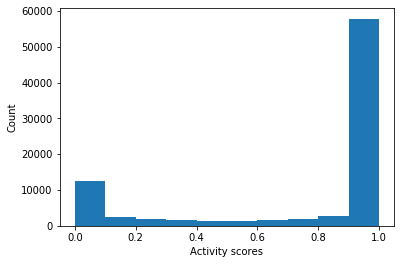
As we can see, most of the molecules generated by the model are predicted as active, although there are a few molecules that have been indeed labeled inactive. Furthermore, we see that the model is quite binary and labels most thing as either active or inactive, with few molecules receiving intermediate activity scores.
Uniqueness
Although close to 60% of the structures generated were predicted to be active, at the moment there are still many repeats found in the set of generated molecules. Let’s calculate how many actives the model generated after filtering out repeat molecules. We can do this by running the cell below.
# get only the unique actives
!sed -i "/SMILES/d" output_DRD2_actives/analysis/epochGEN100_validonly_actives.smi # remove any header in the file
!sort output_DRD2_actives/analysis/epochGEN100_validonly_actives.smi | uniq -u > output_DRD2_actives/analysis/epochGEN100_unique_actives.smi
# compute some stats
all_smi = "output_DRD2_actives/analysis/epochGEN100.smi"
total_n_samples = len(open(all_smi).readlines())
all_actives_smi = "output_DRD2_actives/analysis/epochGEN100_validonly_actives.smi"
actives_n_samples = len(open(all_actives_smi).readlines())
unique_actives_smi = "output_DRD2_actives/analysis/epochGEN100_unique_actives.smi"
unique_actives_n_samples = len(open(unique_actives_smi).readlines())
percent_of_actives = unique_actives_n_samples/actives_n_samples * 100
percent_of_all_generated = unique_actives_n_samples/total_n_samples * 100
print(f"Unique actives sampled: {unique_actives_n_samples}")
print(f" * Unique actives make up {percent_of_actives:.1f}% of all actives generated")
print(f" * Unique actives make up {percent_of_all_generated:.1f}% of all molecules generated (including invalid and repeat structures)")
Unique actives sampled: 34886
* Unique actives make up 72.4% of all actives generated
* Unique actives make up 34.6% of all molecules generated (including invalid and repeat structures)
Novelty
Approximately 35% of all the generated molecules are unique actives! But, how many of them are novel? That is, how many of them were not previously seen by the model? For this, we need to calculate how many of the predicted actives appear in the training, testing, and validation sets, respectively:
# first define a few functions that we will use multiple times
def find_common(a, b, label):
""" Find the common SMILES between two lists of SMILES.
"""
common = list(set(a).intersection(set(b)))
n_common = len(common)
percent_common = n_common/len(b) * 100
print(f"Number of molecules reproduced from {label} set: {n_common} ({percent_common:.1f}%)")
return common
def load_SMILES_list(smi_file):
""" Load SMILES from file into a list containing SMILES.
"""
mols = SmilesMolSupplier(smi_file, sanitize=True, titleLine=False)
smiles_list = [Chem.MolToSmiles(mol) for mol in mols]
return smiles_list
# load generated SMILES as a list
generated_smi_file = "output_DRD2_actives/analysis/epochGEN100_unique_actives.smi"
generated_SMILES = load_SMILES_list(generated_smi_file)
# load training, testing, and validation set SMILES as lists
train_smi_file = "data/DRD2_actives/train.smi"
train_SMILES = load_SMILES_list(train_smi_file)
test_smi_file = "data/DRD2_actives/test.smi"
test_SMILES = load_SMILES_list(test_smi_file)
valid_smi_file = "data/DRD2_actives/valid.smi"
valid_SMILES = load_SMILES_list(valid_smi_file)
# find common structures between the generated and {training/testing/validation} set
common_train = find_common(generated_SMILES, train_SMILES, label="training")
common_test = find_common(generated_SMILES, test_SMILES, label="testing")
common_valid = find_common(generated_SMILES, valid_SMILES, label="validation")
# combine all three splits
all_DRD2_SMILES = train_SMILES + test_SMILES + valid_SMILES
# find common structures between the generated set and the entire set of known DRD2 actives
common_entire_set = find_common(generated_SMILES, all_DRD2_SMILES, label="entire")
# remove the common structures and save only the NOVEL actives
novel_mols = []
for smiles in generated_SMILES:
if smiles not in common_entire_set:
novel_mols.append(Chem.MolFromSmiles(smiles))
novel_smi_file = generated_smi_file[:-4] + "_novel.smi"
writer = SmilesWriter(novel_smi_file)
for mol in novel_mols:
writer.write(mol)
# compute some stats
n_novel = len(novel_mols)
percent_novel_of_actives = n_novel/len(generated_SMILES) * 100
percent_novel_of_all = n_novel/total_n_samples * 100
print(f"Total novel actives generated: {n_novel}")
print(f" * Novel actives make up {percent_novel_of_actives:.1f}% of unique actives generated")
print(f" * Novel actives make up {percent_novel_of_all:.1f}% of all molecules generated (including invalid and repeat structures)")
Number of molecules reproduced from training set: 2157 (62.6%)
Number of molecules reproduced from testing set: 42 (9.7%)
Number of molecules reproduced from validation set: 50 (11.6%)
Number of molecules reproduced from entire set: 2249 (52.2%)
Total novel actives generated: 13231
* Novel actives make up 37.9% of unique actives generated
* Novel actives make up 13.1% of all molecules generated (including invalid and repeat structures)
As we can see, roughly 13% of all structures generated are predicted to be novel, unique actives (i.e. not known actives)!
Furthermore, we can see that roughly 10% of the testing set actives were reproduced by the generated set, and roughtly 10% of the validation set. This is a nice validation for our model, as it shows how the trained GGNN is capable of generating true actives that it has never seen before, and gives us confidence that the novel actives generated are also likely true actives.
Let’s visualize some of the novel actives with a predicted activity score of 1.0:
import random
from rdkit.Chem.Draw import MolsToGridImage
# filter novel actives with an activity score < 0.999
find_actives(smi_file="output_DRD2_actives/analysis/epochGEN100_unique_actives_novel.smi",
activity_threshold=0.999,
verbose=False)
# filtered actives have been written to the file below, which we will use to plot the novel highest-scoring actives
smi_file = "output_DRD2_actives/analysis/epochGEN100_unique_actives_novel_actives.smi"
# load molecules from file
mols = SmilesMolSupplier(smi_file, sanitize=True, nameColumn=-1)
n_samples = 24
mols_list = [mol for mol in mols]
mols_sampled = random.sample(mols_list, n_samples)
png_filename=smi_file[:-3] + "png" # name of PNG file to create
labels=list(range(n_samples)) # label structures with a number
# draw the molecules (creates a PIL image)
img = MolsToGridImage(mols=mols_sampled,
molsPerRow=6,
legends=[str(i) for i in labels])
img.save(png_filename)
# display
from IPython.display import Image
Image(filename=png_filename)
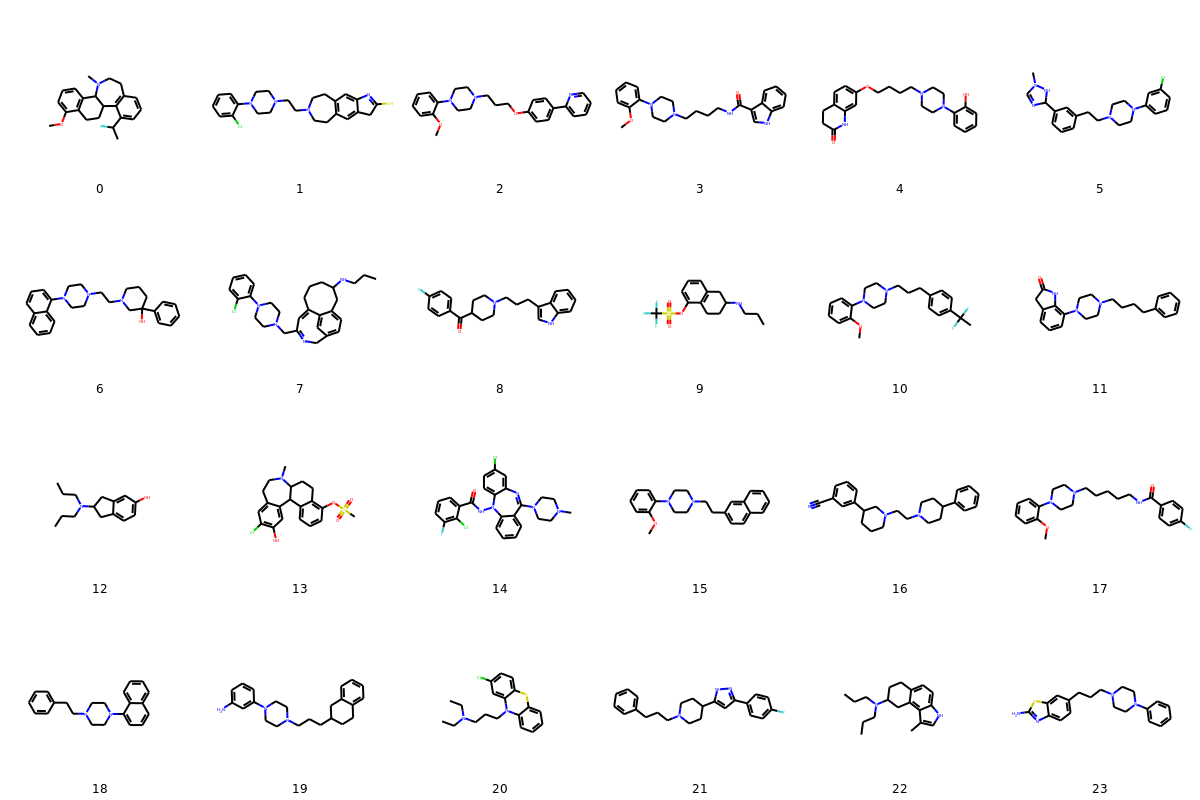
The molecules look great.
As a result, we have successfully used GraphINVENT to predict novel DRD2 actives! The next step would be to of course confirm the activity of these molecules using more accurate methods, such as synthesis and experimental verification, but for those of us who are constrained to building molecules on a computer, this is pretty satisfying.
Summary and conclusions
Using GraphINVENT, a public dataset, and a published QSAR model, we have shown you how one can generate novel predicted actives against the DRD2 target. The calculations for this blog post take about 2.5 hours to run on a GPU (including the analysis), illustrating the speed with which one can design and analyze promising new molecules for an interesting drug discovery task.
Not only have we generated new molecules and predicted their activities, but also validated that the molecules generated are likely to be actives, as approximately 10% of the testing set and 10% of the validation set, which are not seen during training, were regenerated by our trained model (using 100,000 samples). Furthermore, approximately 40% of the unique set of generated structures was molecules which were not seen before in either the training, testing, and validation set; this comprised of 13,231 promising new molecules to explore further.
Of course, it goes without saying that the process of drug discovery is not simple. In a real drug discovery project, one would need to further analyze the viability of generated molecules using both additional modeling tools and, more importantly, experiments. Demonstrating target activity is just one facet of the complicated drug discovery landscape that includes things like improving a molecule’s ADMET properties, synthesizing the molecules in a lab, etc.
The GraphINVENT platform has been made publicly available on GitHub so that it can continue to be explored for molecular design applications. Based on the modular nature of the code, models can be easily added to or modified in GraphINVENT, such that it is easy to investigate different message passing, message update, or graph readout functions, as well as different global readout functions. Furthermore, it is also easy to investigate new datasets for molecular generative tasks, with some limitations. If you would like to use GraphINVENT generative models in your own work, we recommend you check out the tutorials available in the repo for further details.
String-based generative models are currently the state-of-the-art for molecular generation due to the speed and accuracy with which they train on and generate new molecules. However, I believe that graph-based methods are the future of molecular design, as despite the greater computational expense associated with graphs, they are a more natural (and flexible) representation for representing molecules. They can also train well on relatively few examples of molecules. GraphINVENT is just one of many recent examples of promising graph-based methods for molecular design, and as the field becomes more mature, I am sure that many of the issues currently facing graph-based methods will be addressed, making it possible to extend graph-based generative models to larger molecules and more complex generation tasks (e.g. 3D modeling!).
Hope you find this useful and as exciting as I do!
Rocío
References
Generative model:
- https://github.com/MolecularAI/GraphINVENT
- Mercado et al. Graph Networks for Molecular Design. ChemRxiv preprint. 2020. link
- Mercado et al. Practical Notes on Building Molecular Graph Generative Models. ChemRxiv preprint. 2020. link
Pre-trained QSAR model:
- https://github.com/pcko1/Deep-Drug-Coder
- Kotsias et al. Direct steering of de novo molecular generation with descriptor conditional recurrent neural networks. Nature Machine Intelligence. 2020. link
Original DRD2 dataset:
Pingback: Cambridge Cheminformatics Newsletter, 9 November 2020 – DrugDiscovery.NET – AI in Drug Discovery
Thank you very much for a great post. I am trying to use my dataset and when I execute the code below
“`
import os
from submit import write_input_csv
# create the output directory
data_path_minus_data = data_path[:-5]
dataset_output_path = f”{data_path_minus_data}output_{dataset}”
dataset_output_path = os.path.join(dataset_output_path, jobname)
print(f”* Creating dataset directory {dataset_output_path}/”)
os.makedirs(dataset_output_path)
# specify the output directory in `params`
params[“job_dir”] = f”{dataset_output_path}/”
# write the `input.csv` file
write_input_csv(params_dict=params, filename=”input.csv”)
# submit the job (using the command line)
!python graphinvent/main.py –job-dir output_DRD2_actives/preprocessing_example/
“`
I get the following error:
“Exception: Input B not in allowable set [‘C’, ‘N’, ‘O’, ‘F’, ‘S’, ‘Cl’, ‘Br’]. Add B to allowable set in `features.py` and run again.”
Could you please indicate the location of `features.py` or I need to create it myself?
Thank you.
Hi Vitali, It seems that you have boron atoms in your dataset, and you need to extend the list of atom-types. Rocío, correct me if I’m wrong, this is now done by adding the information to the params dictionary, e.g in the preprocessing script? where you need to expand the atom-list like this?
params = { “atom_types”: [“C”, “N”, “O”, “F”, “S”, “Cl”, “Br”, “B”] …
Yes, what Esben wrote is correct, you can update it in “submit.py” file by adding “B” to the atom types e.g. defining:
“atom_types”: [“C”, “N”, “O”, “F”, “S”, “Cl”, “Br”, “B”]
It appears I need to update that error message, so thank you for pointing this out.
Thanks for the clarification 😀
Thank you for the detailed tutorial. I followed the example and data you provided for preprocessing, but I encountered the error message “NameError: name’hyperparameters’ is not defined”. Could you please explain the reason for the error?
Thanks for the interest in using GraphInvent. I’m not sure at the source of your error, could you provide a bit more details? At what step do you encounter the message, and please provide the full error message.
When I tried to use the submit command “python graphinvent/main.py –job-dir output_DRD2_actives/preprocessing_example/” with v2.0,an error message “NameError: name’hyperparameters’ is not defined” appeared. The full error message was as follows.
“File ‘GraphINVENT-master/graphinvent/parameters/defaults.py'”, line 362, in
parameters.update (hyperparameters)
NameError: name’hyperparameters’ is not defined”
I also tried v1.0, but there was the following problem.
“File ‘GraphINVENT-master/graphinvent/parameters/defaults.py'”, line 383, in
params_dict.update (model_specific_hp_dict)
NameError: name ‘model_specific_hp_dict’ is not defined.”
Hi Cindy – I am not sure for the source of the error, as I am not able to reproduce it from the information you provided. Feel free to email me and I can help you figure it out – I have made some recent changes to the code (pushed a new version to the repo), and the new version may no longer be 100% backcompatible with the instructions I have shown on the blog. I updated the tutorials available in the repo but it is of course possible I missed something, let me know if this is the case!
The easiest way to run the code is using the provided submission script (`submit.py`, where you can specify the dataset to use as well as the hyperparameters). Once you have updated the submission script you can run it from within the graphinvent conda environment using “python submit.py”. However, if this is not working for you, please reach out to my email (rocio.mercado [at] astrazeneca [dot] com) and I can help you figure it out. It appears that when you try to run graphinvent there is an issue with the paths, as it cannot find the hyperparameters (I am guessing, not sure though).
Thank you so much for your detail blog!
I met the same errors when I ran it on Jupyter notebook. When I ran the following cell
import os
from submit import write_input_csv
# create the output directory
data_path_minus_data = data_path[:-5]
dataset_output_path = f”{data_path_minus_data}output_{dataset}”
dataset_output_path = os.path.join(dataset_output_path, jobname)
print(f”* Creating dataset directory {dataset_output_path}/”)
os.makedirs(dataset_output_path)
# specify the output directory in `params`
params[“job_dir”] = f”{dataset_output_path}/”
# write the `input.csv` file
write_input_csv(params_dict=params, filename=”input.csv”)
# submit the job (using the command line)
!python graphinvent/main.py –job-dir output_DRD2_actives_new/preprocessing_example/
I got:
* Creating dataset directory ./output_DRD2_actives_new/preprocessing_example/
Traceback (most recent call last):
File “graphinvent/main.py”, line 5, in
import util
File “/home/xzhang/projects/GraphINVENT/graphinvent/util.py”, line 14, in
from parameters.constants import constants
File “/home/xzhang/projects/GraphINVENT/graphinvent/parameters/constants.py”, line 14, in
import parameters.defaults as defaults
File “/home/xzhang/projects/GraphINVENT/graphinvent/parameters/defaults.py”, line 364, in
parameters.update(hyperparameters)
NameError: name ‘hyperparameters’ is not defined
However, if I used $python submit.py from the terminal, it worked.
Hi Zhang,
Thanks for your interest. I think Rocío made some updates and refactoring of the code on the github repository, so the blog may not be accurate anymore. Please try and follow the documentation from the github repository about how to setup the hyperparameters.
Best Regards
Esben
Thank you for your great article!
When I used the following parameters to generate new molecules,
params = {
“atom_types”: [“C”, “N”, “O”, “F”, “S”, “Cl”, “Br”],
“formal_charge”: [-1, 0, +1],
“max_n_nodes”: 72,
“job_type”: job_type,
“dataset_dir”: f”{data_path}{dataset}/”,
“min_rel_lr”: 1e-3,
“model”: “GGNN”,
“lrdf”: 0.9999,
“lrdi”: 1000,
“sample_every”: 10,
“batch_size”: 500,
“init_lr”: 1e-4,
“epochs”: 100,
“block_size”: 50000,
“generation_epoch”: 100,
“n_samples”: 100000,
}
I got 200 files named epochGen*.smi and each file contains 100,000 or more molecules. Because n_samples is 100,000 and batch_size is 500, so I have 200 batches. However, I don’t understand why each file has 100,000 new molecules. The same thing happened when I tried gdb13_1k dataset following your tutorials. Could you please explain it for me? Many thanks
Hi Zhang,
Thanks for your question. I would think that it’s the generation done after each epoch. However, you specifify 100 epochs, so the number of 200 doesn’t fit that. Maybe I can get Rocío to clarify.
Best Regards
Esben
Hi Zhang,
This is actually a bug, I had noticed it before and believe I fixed it in the latest version. It was a small mistake in calculating the # of batches needed to get the correct number of samples (division by the wrong variable) in the method `generate_graphs()`.
Do you know, approximately, the date that you cloned the code, or can you confirm that you are using the most up-to-date version of GraphINVENT (last changes made in June)?
In the version you have, the code is simply generating too many samples (by mistake), so you have generated 20,000,000 molecules. If you want 100,000, you can take any subset.
Thank you for bringing this to our attention.
Rocío
Thank you for your reply. I just git clone your repo last week from your GitHub page.
Hello,
Thanks for writing a detailed blog post. With regards to evaluation mentioned in the paper, I have a doubt regarding the target APD. Since generative models usually don’t have a ground truth, how are you defining the values of target APD? Could you please elaborate?
Best,
K.Vikraman
Hi Virkaman, thanks for the interest in GraphINVENT and for asking. The action probability distribution is learned from training on many explicit examples. We break down existing molecules into smaller graphs, by imagining the kind of steps that are needed to build the target graph and use these as the ground truth for the training. Let me try and explain with some simple examples.
We have the molecule 1-propanamin CCCN, the actions should be start with C, add C to 1, add C to 2, add N to 3, stop. Thus in the training the neural network would see the graphs and the actions
C – Add C to 1
CC – Add C to 2
CCC – Add N to 3
CCCN – Stop
Similarly for Butane we could have
C – Add C to 1
CC – Add C to 2
CCC – Add C to 3
CCCC – Stop
If we only trained on these samples, the APD after first C would get to be 100% add C to -1, and the same for CC, But for the last addition step, both using the graph CCC as input, the APD learned would be 50% Add C to 3, and 50% Add N to 3.
Of course in GraphInvent, there are many more examples with more complex graphs paired together with the next action in the training set, giving rise to more complex APD. We also examined the differences in breadth first or depth first for the graph disconnections. https://pubs.acs.org/doi/pdf/10.1021/acs.jcim.1c00777 (there’s also a preprint on ChemRxiv).
Thank you for your great article!
When I used the following parameters to generate new molecules,
import os
from submit import write_input_csv
# create the output directory
data_path_minus_data = data_path[:-5]
dataset_output_path = f”{data_path_minus_data}output_{dataset}”
dataset_output_path = os.path.join(dataset_output_path, jobname)
print(f”* Creating dataset directory {dataset_output_path}/”)
os.makedirs(dataset_output_path)
# specify the output directory in `params`
params[“job_dir”] = f”{dataset_output_path}/”
# write the `input.csv` file
write_input_csv(params_dict=params, filename=”input.csv”)
# submit the job (using the command line)
!python graphinvent/main.py –job-dir output_DRD2_actives/preprocessing_example/
I got: cannot import name ‘write_input_csv’ from ‘submit’
where is “write_input_csv’ info?
Hi, Unfortunaly I first see this comment now, as the notification went to the spam folder. Have you figured out the issue? The error tells that it can’t import the function write_input_csv from the submit module, where I suspect that the interpreter can’t find the submit module. I think Rocío may be able to help you better