Learn how to improve SMILES based molecular autoencoders with heteroencoders
Earlier I wrote a blog post about how to build SMILES based autoencoders in Keras. It has since been a much visited page, so the topic seems interesting for a lot of people, thank you for reading. One thing that I worried about was how nearby molecules seem more related when comparing the SMILES strings rather than the molecules. I’ve done some more investigations and come up with a possible solution in the form of heteroencoders. The details are already available in a preprint on Arxiv.org or read on below for an overview.

Galileo Galilei 1564-1642
Galileo Galilei is quoted for saying:”Measure what is measurable, and make measurable what is not so”, so I tried to quantify the equality of the latent space to the SMILES similarity using ideas from biological sequence alignment and scoring. It turned out to be quite easy with the Biopython package.
# Import pairwise2 module
from Bio import pairwise2
def seq_similarity(smi1, smi2):
alignments = pairwise2.align.globalms(smi1, smi2,1,-1,-0.5,-0.05)
return alignments[0][2]
The function aligns the two SMILES sequences using dynamic programming and returns the score, which is a sum of +1 for each character match, -1 for character mismatch, -0.5 for opening a gap and -0.05 for extending a gap. The scores were then computed between a single molecule and 1000 others from the test set with a list comprehension.
seq_sims = [seq_similarity(rootsmiles, smile) for smile in test_smiles.values[0:1000]]
These values were then plotted against the Euclidean distance in the latent space between the same molecules after log conversion and the correlation coefficient was used as a measure of the equivalence between the two spaces. There may be smarter ways to compare equality of spaces, but this approach got the job done.
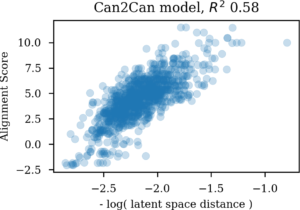
Comparing molecular similarities as SMILES alignment scores with latent space Euclidian distances
A similar approach can be used to compare the latent space with the Morgan similarity between the molecules. Here also for the canonical SMILES to canonical SMILES autoencoder.
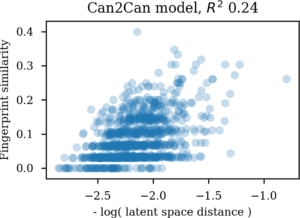
Comparing molecular similarities as Morgan tanimoto coefficients with latent space Euclidian distances
For the autoencoder the SMILES sequence space seem much more related to the latent space than the Morgan similarity based one. So there indeed seem to be more SMILES than molecules in the latent space for autoencoders. Likewise, if an autoencoder is challenged with enumerated SMILES, that is different versions of non-canonical SMILES strings for the same molecule, they ends up in very different areas of the latent vector space. This gives me serious doubts about its usefulness for navigating chemical space.
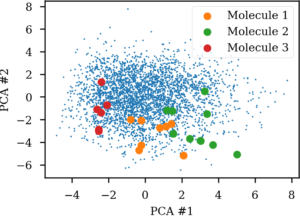
Latent space projection of 10 different SMILES forms of three molecules.
A Possible Solution: Use Heteroencoders!
So what is a heteroencoder? Its simply an autoencoder, but where the network is asked to trans-code between different formats, versions or representations of the same molecular entity during training. Combinations could be from a chemical depiction to a SMILES string, or more simple, from one form of the SMILES string of a molecule to another form of the SMILES string of that molecule (SMILES enumeration). I’ve previously written about SMILES enumeration and how it can be used to work as data augmentation for chemical learning, so we already know how to get multiple different SMILES forms of the same molecule. There seem actually to exists many more heteroencoders than autoencoders, which may be the special case. The concept is illustrated in the figure below.
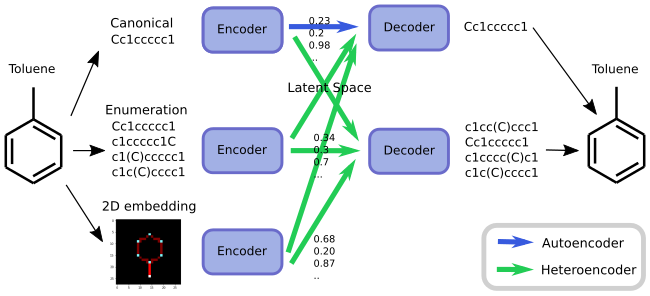
But “hey”, one could say, “wouldn’t translating from a random SMILES form to another random SMILES form be like translating from a random language to another random language? That can’t possibly work!”.
The analogy is probably somehow right, and I thought so too for a long time. But artificial neural networks just keep surprising me, so ended up trying anyway. Now I think that the teacher forcing used during training of the decoder is very important for the success of the approach. Teacher forcing is a way to help the recurrent neural networks train efficiently on longer sequences. As the prediction is done character by character (or word by word for word embeddings), the error is calculated after each character, and the recurrent neural network corrected by the correct character or word by the “teacher”. So mistakes made early in the sequence will not automatically ruin the character by character predictions later in the sequence.
If we stay in the language analogy, I may ask the network to translate the English sentence “Hello, how are you today?” into another language. After encoding the sentence into a latent vector in the bottleneck layer, the networks decoder tries the first word in German “Guten”, but is immediately told that it was wrong and that the correct is “Goddag” (Danish for hello). The next word will thus probably not be the guess “Tag” in German, but more likely a continuation of the Danish sentence “Goddag, hvordan går det?”. In recurrent neural networks the following computations and predictions will be influenced by the previous computations and predictions, so the teachers correction of the words will influence the decoding of the rest of the words. It’s quite fascinating that it works. As I used customized generator objects to train the networks, it is quite unlikely that they ever saw the same pair of enumerated SMILES during training under the enum2enum regime.
SMILES strings are luckily much simpler than language. The figure below compares how it looks when an autoencoder (A) or a heteroencoder (B) decodes the latent representation of a molecule. The figures show as a heat map how the probability was for prediction the next characters for each step of the decoding going left to right.
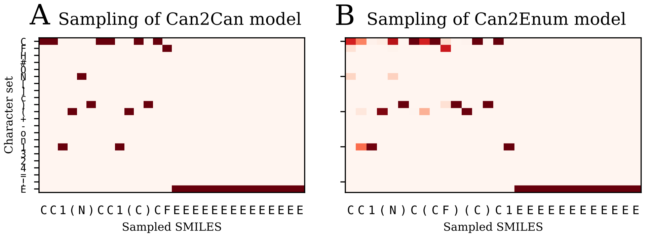
The autoencoder on the left (A) is completely certain how to decode each character and produces both the correct molecule and the correct canonical SMILES string each time. In contrast, the predictions of the heteroencoder to the right (B) are more fuzzy. The first character predicted is most likely a carbon “C”, but there is also a slight chance that the network would decode “N” or “F”. For the first character it samples the probability distribution and decodes “C”. Decoding of the next character is then influenced by this choice. Had the decoder sampled “N” as the first character, the rest of the decoding heat map would look completely different. The decoding of the next character also have a couple of options, but the third one gets fixed as a ring start “1”, probably because of the choice of the first two “CC” only leaves one opportunity for the third. In the end the encoder actually end up decoding a random SMILES string of the right molecule. Quite fascinating. If the decoder was asked a to decode the molecule again, it would end up with a different heatmap due to the choices taken in each decoding step and a different SMILES string.
Practical implications for QSAR and generative de-novo design
So whats the big deal? Using heteroencoders you can decode a molecule into a lot of different SMILES? How’s that going to help anything? It turns out that the change in the task during trainings gives a change in the latent representation. The molecules from the SMILES enumeration challenge above ends up in the same areas in the latent space. The SMILES and Morgan FP similarity correlations balances out in the heteroencoder. But more importantly, the latent space also seem much more relevant as descriptors of the encoded molecules.

Boris Sattarov. MSc Cheminformatics
A bright young scientist I worked together with, Boris Sattarov, expanded the networks to handle ChEMBL sized molecules. He then investigated how latent vectors produced by the networks after different training regimes influenced their usage for QSAR modelling. The results are quite striking. Usage of heteroencoder derived latent vectors works much better than both autoencoders and baseline models built on ECFP4 fingerprints in a range of QSAR tasks for both bioactivity and physico-chemical properties. The difference between the different heteroencoders tested is small, but there is a slight tendency for the networks trained from enumerated SMILES to enumerated SMILES to work the best on average.
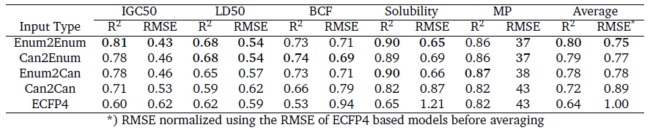
QSAR performance on the held-out test for ECFP4, autoencoder and heteroencoder derived descriptors.
Good ideas usually spring up multiple places in parallel and a group of scientist at Bayer also found that using heteroencoders improved the usability of the latent space for QSAR applications. This gives further confidence in the observations.
However, the choice of training of the decoder also have a marked influence on the molecules produced during decoding. Not only do the decoder produce different SMILES forms of the encoded molecule, it also directly produces novel molecules which may be interesting in de-novo generation and design of compounds. It is possible to encode a single lead compound into a latent vector and sample around it. The decoder gets a bit more “creative” after training on enumerated targets, which may or may not be wanted depending on the applications and wanted outcome. The greater relevance of the latent space as QSAR descriptors should in principle also make the sampled novel molecules more relevant with regard to similar bioactivity and physco-chemical properties. I hope to be able to investigate the practical use of this in the near future.
Recommendations
So if you work with SMILES based molecular autoencoders, I really encourage you to use heteroencoders instead. If deterministic and precise decoding is needed, at least use an enumerated to canonical heteroencoder. If you can live with, want greater uncertainty in the decoding, or are interested in using the latent vectors for QSAR applications, use an enumerated to enumerated heteroencoder.
Read further in the preprint: Improving Chemical Autoencoder Latent Space and Molecular De novo Generation Diversity with Heteroencoders or feel free to leave a comment/question below.
Best Regards
Esben
Pingback: Master your molecule generator: Seq2seq RNN models with SMILES in Keras | Wildcard Pharmaceutical Consulting
How can we use heteroencoders if there is no link to the code? 😉
Could you please share a repo?
Thanks for the interest. There’s the enumeration/randomization capacity in the molvecgen package: https://github.com/EBjerrum/molvecgen/tree/master/molvecgen. You will probably need to customize the sequence or generator object to work with your deep learning framework.
If you want a more full package, the DDC code we released has this capacity: https://github.com/pcko1/Deep-Drug-Coder
Hello! I’m a student and trying to implement similar heteroencoder, however, I don’t understand how loss is calculated in case of can2enum version. It’s pretty tricky for me because how should we handle with multiple outputs for one canonical input?
Thanks for the interest. To address your question, there is NO difference in loss function. You use a SoftMax and categorical cross-entropy, exactly similar to this blog-post: https://www.cheminformania.com/master-your-molecule-generator-seq2seq-rnn-models-with-smiles-in-keras/. What’s important is that you have teachers forcing. The loss will not go as low as in the X2can cases, but that’s because the output probability distributions will be handling multiple output possibilities for the characters. See Figure 10 in this publication: https://arxiv.org/pdf/1806.09300.pdf, which indicates how the network uses the feedback from the sample to choose between several different possibilities of the right molecule during sampling.
I would recommend to either use a Enum2can or a enum2enum. Can2enum will not be generalizing as good and be as robust as enum2enum, and you’ll still have the issues of handling multiple output for the same molecules during sampling.
You can also take a look at the implementation in the deep drug coder project, https://github.com/pcko1/Deep-Drug-Coder, which can both handle heteroencoders and cRNNs. https://www.nature.com/articles/s42256-020-0174-5 or https://chemrxiv.org/articles/Direct_Steering_of_de_novo_Molecular_Generation_using_Descriptor_Conditional_Recurrent_Neural_Networks_cRNNs_/9860906/2
Thank you so much for fast and comprehensive response! Actually, I’m scrutinizing your article (“A de novo molecular generation method using latent vector based generative adversarial network”) and I misread the string (previuosly thought that the mentioned error was using during training, but now I get it!):
You’re welcome. Go ahead and try it out, look at the output from the different layers with plots/heat-maps, both before and after training. Sometimes that can really help with understanding. Good luck and happy training.
OMG! What a great and inspiring paper to read! I have never heard about heteroencoders before until I read you paper
Thanks for commenting. The paper was the first one with the concept. To be fair, Okko’s group from Bayer independently worked on a similar concept and published shortly after me and Boris.
I tried to google search with several keywords of Okko’s group from Bayer independently, but I could not find his paper. Could you mind if I can ask you to comment it here, please?
Ah, OK, here’s the link for their code where they also have a reference https://github.com/jrwnter/cddd