How to solve problems with coordinate bonds in Rdkit
 When I have been working with chemical databases and import of molecules I have encountered numerous problems with the way chemical structures are drawn. Most often the problem arises as creative users in the past have had a problem with registering a compound the way they wanted it. Sometimes the used software did not support a correct way, so the users found a workaround, which can give rise to errors with other software packages. A common problem arises with organometallic compound and coordinate bonds. Read on if you want to learn more about how to handle these kind of compounds with Rdkit.
When I have been working with chemical databases and import of molecules I have encountered numerous problems with the way chemical structures are drawn. Most often the problem arises as creative users in the past have had a problem with registering a compound the way they wanted it. Sometimes the used software did not support a correct way, so the users found a workaround, which can give rise to errors with other software packages. A common problem arises with organometallic compound and coordinate bonds. Read on if you want to learn more about how to handle these kind of compounds with Rdkit.
Chemical Bond Types and File Formats
Chemical bonds come in a variety of versions. A regular covalent bond is formed when two atoms combine orbitals with only one electron each. This gives a molecular orbital with two electrons. However, if the electronegativity differences are large enough, a ionic bond is formed where the most electronegative atom attracts both electrons. Both these situations are handled by Rdkit without problems.
For the coordinate or dative bond, which this blog post is about, one atom donates both electrons into a covalent bond forming with another atom possessing an empty orbital. These differences in bond types can give rise to problems with software and formats that only supports the other bond types. The widely used .mol and .sdf formats didn’t contain dative or coordinate bonds in the V2000 format, but from around 2011 the hydrogen bond and dative bond was added to the specification for the V3000 format. You can read more about the formats and specification at https://en.wikipedia.org/wiki/Chemical_table_file and http://download.accelrys.com/freeware/ctfile-formats/ctfile-formats.zip (Sorry, the last file is behind a Login wall).
I recently had a need to work with database registration of chemical complexes, where it could be nice to specify the coordinate bonds. In the existing .sdf files they were specified as regular covalent bonds, but this led to problems with the internal chemistry model and electron accounting in both RDKit and Chemdraw. MarvinSketch supports a coordinate bond type and can read and write correctly formed v3000 Molfiles, ChemDraw version 15 had some issues with the V3000 format that I have informed Perkin-Elmer about. I don’t know if they have fixed them.
The bond type are defined in the BONDS section of the v3000 molfile in the second column of the bond line (M V30 ). Below is shown a snippet where the bond type 9 is the dative or coordinate bond type.
M V30 BEGIN BOND M V30 1 1 1 2 M V30 2 1 2 3 M V30 3 1 1 4 M V30 4 9 4 5 M V30 5 9 3 5 M V30 6 1 3 6
As an example I have chosen DiSodiumCopper-EDTA, a beautiful blue compound. Theres a video on YouTube showing how copper can make different coordination compounds resulting in color changes. https://www.youtube.com/watch?v=deNWxchzDRg. EDTA is a chelating agent capable of forming multipe coordination bonds, there is much more information about it on Wikipedia: https://en.wikipedia.org/wiki/Ethylenediaminetetraacetic_acid
RDKit and Dative/Coordination Bonds
Internally RDKit has support for a dative bond type, but after experimenting a bit, it was clear that v3000 formatted mol files with dative bonds could not be loaded into RDkit. If the bond types where changed to dative in an RDKit python session, they could also not be saved into .mol format when forcing the v3000 format.
Luckily, RDKit is open source (Thanks Greg and others :-), so it was possible to dive into the code and see the import and export functions. The C++ function responsible for loading mol files in v3000 formats was found in the ParseV3000BondBlock function in the Code/GraphMol/FileParsers/MolFileParser.cpp file. There I found a switch case handling the bond types and it was easy to add two extra cases for bond type 9 and 10.
case 9:
bond = new Bond(Bond::DATIVE);
break;
case 10:
bond = new Bond(Bond::HYDROGEN);
break;
For saving the dative bonds in V3000 formats, the bond types are handled in the GetV3000BondCode function in the /Code/GraphMol/FileParsers/MolFileWriter.cpp file.
case Bond::DATIVE:
res = 9;
break;
case Bond::HYDROGEN:
res = 10;
break;
The complete patch file is shown in the end of this blog post.
Test drive of import, handling and export of coordinate bonds
Let’s see what happens when we try to work with molecules with Rdkit in a Python session 🙂
from rdkit import Chem
from rdkit.Chem import Draw
cuEDTA = Chem.MolFromMolFile('DisodiumCopperEDTA_v3000.mol')
Draw.ShowMol(cuEDTA)
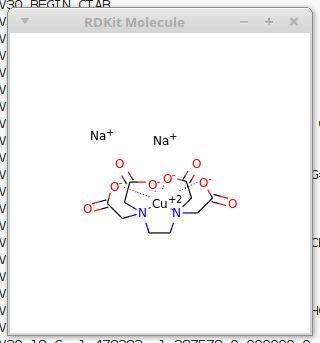
CuEDTA rendered by RDkit
Nice, Coordinate bonds are shown with dotted lines.
Chem.MolToMolFile(cuEDTA,'Testsave.mol',forceV3000=True)
This also gives the right bond type in the Testsave molfile (M V30 4 9 4 5 ). Good. This illustrates how easy it sometimes is to work with the RDKit codebase. I’ve just started to work with the CPP code of RDKit and I have not done extensive testing of the patch yet. However, All the ctest tests included with the RDKit source completes without error, so it seem no to have broken anything.
Cheers,
Esben
The patch file to enable v3000 support in RDkit:
diff --git a/Code/GraphMol/FileParsers/MolFileParser.cpp b/Code/GraphMol/FileParsers/MolFileParser.cpp
index 1ca663b..3b4f9e2 100644
--- a/Code/GraphMol/FileParsers/MolFileParser.cpp
+++ b/Code/GraphMol/FileParsers/MolFileParser.cpp
@@ -1963,6 +1963,12 @@ void ParseV3000BondBlock(std::istream *inStream, unsigned int &line,
bond = new Bond(Bond::AROMATIC);
bond->setIsAromatic(true);
break;
+ case 9:
+ bond = new Bond(Bond::DATIVE);
+ break;
+ case 10:
+ bond = new Bond(Bond::HYDROGEN);
+ break;
case 0:
bond = new Bond(Bond::UNSPECIFIED);
BOOST_LOG(rdWarningLog)
diff --git a/Code/GraphMol/FileParsers/MolFileWriter.cpp b/Code/GraphMol/FileParsers/MolFileWriter.cpp
index 793c09f..957ffac 100644
--- a/Code/GraphMol/FileParsers/MolFileWriter.cpp
+++ b/Code/GraphMol/FileParsers/MolFileWriter.cpp
@@ -920,6 +920,12 @@ int GetV3000BondCode(const Bond *bond) {
case Bond::AROMATIC:
res = 4;
break;
+ case Bond::DATIVE:
+ res = 9;
+ break;
+ case Bond::HYDROGEN:
+ res = 10;
+ break;
default:
res = 0;
break;
Hi,
I am a novice coder and am trying to generate some descriptors for metal complexes for machine learning. My complexes contain two dative bonds between a nitrogen and the metal centre. I have been able to solve this issue with SMILES by adding ‘->’ to the txt files at points where the dative bonds should be. However, when converting from .sdf (v3000 mol) file I have the same issue of RDKit ERROR: [15:32:43] Explicit valence for atom # 31 N, 4, is greater than permitted. I want to add this fix to my source code but I’m unsure how. Would you be able to contact me and point me in the right direction if possible?
Thanks very much.
Molly
Hi,
Thanks for commenting on my very old blog-post. I think nowadays that RDKit support dative bonds out of the box and there was also a lot of discussion about the extension of the SMILES format at the last UGM, but I haven’t used dative bonds recently. I think your problem is not related to your dative bonds, but rather that you have some tetravalent nitrogen atom in the SDF which is not charged. Check this blogpost on tips how to read in the SDF without sanitization so that you can at least depict the structure and figure out where your issue is: The Good, the Bad and the Ugly RDKit molecules Also, be aware that many descriptors may not be able to really handle the dative bonds, so please check how your wanted descriptors how they treat them (e.g. ignore them or treat them as ordinary bonds), and think if that is what your want. You may also want to use the other channels of support for RDKit as the mailing list.
Best Regards
Esben
Hi Molly,
I am working on a similar issue with you (metal complexes with ML methods). Do you mind if we can communicate about this? My email is shuaiwang.hku@gmail.com
Thanks and best,
Jerry
I think this blog and the reply to Molly are very useful. I am working on a similar issue with Molly as well (metal complexes).
What you said “Also, be aware that many descriptors may not be able to really handle the dative bonds, so please check how your wanted descriptors how they treat them (e.g. ignore them or treat them as ordinary bonds), and think if that is what your want” is really meaningful.
For instance, I have changed the dative bond manually and it can be seen correctly via rdkit. However, when I use “from rdkit.ML.Descriptors import MoleculeDescriptors” to calculate the descriptors, some things went wrong like the molecular weights (there will be more hydrogen counted into the molecular weight). I thought this series of descriptors may treat dative bond as a simple single bond, but not pretty sure.
Would you mind giving more suggestions or anything we need to take care of when dealing with metal complex and coordinate bonds?
Thanks very much!
Best regards,
Jerry
Yes, exactly, it sound from your example as the bond is not really recognized and there are added more implicit hydrogens to the atoms during descriptor calculation.
I would probably check up on how RDKit handles dative bonds now, as RDKit has been developed further since I needed to make the hack to create the depictions of the dative bonds. Can vanilla RDKit read v3000 molfiles with dative bonds? Does the dative bonds do as expected with respect to the descriptor calculation?
Hi Esben,
Thanks very much for your reply. I would check and see if we can find some valuable and reliable descriptors for the metal complex.
Have a nice day!
Thanks again and best regards,
Jerry