pdChemChain – linking up chemistry processing, easily!
I’ve been working on a project intermittently for some time, and I recently packaged it up and published it on GitHub, hoping it could be useful to others. I have mixed feelings about this release – a bit of excitement tempered with a sense of self-consciousness…

As the title suggests, the project is a Python package called pdchemchain. It’s a tool for processing and analyzing chemical data using Pandas, allowing users to chain together various chemical steps to manipulate the data in the dataframe. It also have some extra features that makes it easier to setup and manage the configuration of the pipeline
So? Why another Pipelining package?
What makes me feel a bit silly is that I’m adding yet another pipeline tool to an already crowded field. It’s reminiscent of that old XKCD cartoon about standards, where the principle seems to apply equally to pipelines. There’s already A TON of pipelining tools, software and packages out there, and it makes me a bit silly to think that I should try to add yet another one to the bunch. I’ve used a few of them in different settings, but to be honest, never tested all of them. The ones I used never really felt quite right for the way I wanted to work. Different needs naturally lead to different pipeline solutions. Some might be designed for specific purposes (like watering potted plants), some are made for flexibility, others for transporting industrial quantities of materials, and some might serve entirely different functions with alternative mediums.

One of my frustrations when working with pipelines has been the sometimes cumbersome setup and definition process. It often involves defining YAML or JSON configurations, followed by testing that might result in failures after an hour of processing at step number ten and row 123,456. This leads to a slow cycle of modification, debugging, and retesting.
Frequently, I found myself loading data for exploratory analysis in a Jupyter or IPython interactive session, only to then need to restructure it into a pipeline format, run it externally, and return to the interactive session to reload the data. It’s not that I dislike pipelines, but I prefer to maintain flexibility, and having multiple interactive sessions with loaded data is crucial for my workflow.
These experiences led me to consider developing a set of reusable components that could quickly assist with common processing tasks on pandas dataframes, without the need to repeatedly write the same functions. While pandas does offer methods for chaining operations on dataframes, I wanted something more interactive and stepwise. Over time, this idea evolved into something more comprehensive.
If you’re interested in a tool that enables pipeline definition in interactive sessions, while also allowing combination of steps and preservation for future server-side and command-line use, continue reading. I believe you might find value in what follows 😉.
Over time I evolved some dogma around some principles for designing the API that works quite well.
{kind=link}
API principle #1. Pandas in – Pandas out

The core principle of this package is straightforward: after instantiating a class, it should be able to process a dataframe and return a dataframe. I’ve termed these classes “Links”. This approach significantly simplifies integration into your existing workflow. It also facilitates testing, as you can easily load a small test dataframe and verify that the component functions as expected, leveraging the rich display capabilities of dataframes in Jupyter notebooks.
I considered alternative designs. For instance, I appreciate the fail exit port feature in components from tools like Knime or Pipeline Pilot. This feature allows your pipeline to process the samples or rows it can while clearly reporting those that failed. However, having some components return one dataframe and others return two introduced considerable complexity in pipeline definition.
Ultimately, I decided to adhere strictly to the pandas in, pandas out principle, despite other options potentially offering more flexibility. This decision was driven by a desire for simplicity and consistency across the package.
Let’s examine a Link in action with a test:
import pandas as pd
from pdchemchain.links.chemistry import MolFromSmiles
link1 = MolFromSmiles()
link1.set_log_level("info") #With debug its a bit too chatty for a blogpost
df = pd.DataFrame({"Smiles": ["CCO", "CCC"]})
df_out = link1(df) # You can also use df = link1.apply(df)
df_out
2024-10-01 10:56:04,865 - INFO - rdkit: Enabling RDKit 2023.09.3 jupyter extensions 2024-10-01 10:56:05,082 - INFO - pdchemchain.links.chemistry.MolFromSmiles: Processing dataframe with 2 rows, row by row
| Smiles | ROMol | |
|---|---|---|
| 0 | CCO | <rdkit.Chem.rdchem.Mol object at 0x7247f871b5a0> |
| 1 | CCC | <rdkit.Chem.rdchem.Mol object at 0x7247f871b610> |
Of course, the previous example could have been accomplished with a few simple RDKit lines, or by using the efficient PandasTools.AddMoleculeColumnToFrame function in RDKit. However, I ask for your patience; having the conversion in a separate link offers some additional benefits that I’ll discuss later in this post.
API principle #2. A Chain is also a Link

What do I mean by “a chain is also a link”? Essentially, the pipeline itself is a special subclass of a link, and it can be utilized in the same manner as any other link. This design allows you to combine chains, add a link to a chain, or incorporate a chain into a link.
This property facilitates a straightforward approach to building your pipeline from its constituent parts, which can be developed and tested individually and interactively. By overloading the dunder method __add__ in the class definition, we enable a natural and arguably Pythonic way of constructing processing chains.
Let’s expand on our previous example by adding a new link:
# Showing the step-wise approach on previous output from pdchemchain.links.chemistry import HeavyAtomCount link2 = HeavyAtomCount() df_out = link2(df_out) df_out
| Smiles | ROMol | HeavyAtomCount | |
|---|---|---|---|
| 0 | CCO | <rdkit.Chem.rdchem.Mol object at 0x7247f871b5a0> | 3 |
| 1 | CCC | <rdkit.Chem.rdchem.Mol object at 0x7247f871b610> | 3 |
# Showcasing building a link and processing the first dataframe chain = link1 + link2 df_out = chain(df) df_out
| Smiles | ROMol | HeavyAtomCount | |
|---|---|---|---|
| 0 | CCO | <rdkit.Chem.rdchem.Mol object at 0x7247f1c60740> | 3 |
| 1 | CCC | <rdkit.Chem.rdchem.Mol object at 0x7247f1c607b0> | 3 |
I find this approach quite straightforward. Python’s operator overloading allows us to add multiple links to a chain, for example:
chain = link1 + link2 + link3
Or even:
chain = sum([link1, link2, link3])
If you already have an existing chain and want to extend it further, that’s also possible:
chain = chain + link1
This design principle significantly enhances the package’s flexibility and modularity, enabling users to construct complex pipelines from simpler components. It fosters an intuitive development process where you can iteratively build and test your pipeline in manageable chunks.
I explored other packages that connect processing steps and configure them in a directed graph, allowing each step to produce output for several other steps. However, with the goal of facilitating quick and easy setup of simple pipelines, I decided against this approach. While there are existing tools that offer this functionality, I found that the increased versatility often came at the cost of increased complexity in pipeline definition.
After creating numerous steps and chaining them together, it can become challenging to maintain a clear overview of your pipeline. Sometimes, you simply need to inspect what you’ve done. What if you forget how you configured link number 1, and now you’ve changed the code of the block, but the object you’re using is from a previous version of the code?
This scenario highlights a common challenge in data science workflows, particularly when working with notebooks. After all, notebooks are the fast-food of data science: nice and fast to write, but not always the best for long-term maintenance of the body – err, I mean code.

API principle #3. All Links are self-documenting
Inspired by scikit-learn’s estimators, which have a .get_params() method that returns a dictionary of the parameters used to configure the estimator, all pdchemchain links were designed with a .get_params() method. This method returns a dictionary of the link’s parameters, making it always possible to check exactly what’s happening in the pipeline. Chains automatically show the configuration of their internal links, so both the individual steps and their specific configurations can be inspected. Let’s see this in action:
chain.get_params()
{'_version_': '0.1.dev1+g93fc090.d20240509',
'_loglevel_': 'INFO',
'_class_': 'pdchemchain.base.Chain',
'links': [{'_class_': 'pdchemchain.links.chemistry.MolFromSmiles',
'in_column': 'Smiles',
'out_column': 'ROMol'},
{'_class_': 'pdchemchain.links.chemistry.HeavyAtomCount',
'in_column': 'ROMol',
'out_column': 'HeavyAtomCount'}]}
The resulting dictionary contains more information than just the parameters (which can be switched off with version=False and log_level=False). This additional information, such as the package version used for the pipeline and the set log level (in this example, “INFO”), is displayed at the top level even though it’s not directly related to the configuration of the outermost link. This extra context can be valuable for debugging and reproducibility.
Below this, we see that the object whose configuration we’re examining is of the Chain class, with a list of two links and their respective configurations. Default values are also shown but can be omitted by setting defaults=False.
Having implemented this self-documentation feature, it was a natural next step to add functionality for saving the configuration to YAML or JSON format. This makes it straightforward to save the pipeline configuration to a file for later inspection, documentation, or to ensure reproducibility. Here’s how you can save and view the configuration:
chain.to_config_file("chain_config.yaml")
!cat chain_config.yaml
__class__: pdchemchain.base.Chain __loglevel__: INFO __version__: 0.1.dev1+g93fc090.d20240509 links: - __class__: pdchemchain.links.chemistry.MolFromSmiles in_column: Smiles out_column: ROMol - __class__: pdchemchain.links.chemistry.HeavyAtomCount in_column: ROMol out_column: HeavyAtomCount
API Principle #4. All links are self-configurable
While documenting the configuration of a link, I realized that it could be equally valuable to reinstantiate a link from its configuration. This concept is similar to the .from_param() method in scikit-learn estimators, which is useful for cloning objects without inheriting any internal state, retaining only the same settings.
Implementing this feature required some recursive function coding due to the nested structure used for the .get_params() method. However, now that it’s operational, I find it incredibly useful. This functionality allows users to easily save a link configuration and reuse it later.
The addition of a command-line interface to run a link from a specified configuration file suddenly gives this package a more enterprise-ready feel. It can now be deployed on servers or cluster nodes to process much larger datasets in parallel, expanding its utility beyond interactive sessions.
Here’s a simple example of how to use this feature:
from pdchemchain import Link
chain2 = Link.from_config_file("chain_config.yaml")
chain2
Chain(links=[MolFromSmiles(in_column='Smiles', out_column='ROMol'), HeavyAtomCount(in_column='ROMol', out_column='HeavyAtomCount')])
More features: Error-handling
Proper error handling is crucial in data processing pipelines. There’s nothing worse than a process failing catastrophically at row number 845,123 in step number 12 of the pipeline. While this has never happened to me (all my pipelines run flawlessly, of course ;-)), I can empathize with how frustrating that would be.
The challenge extends beyond ensuring the pipeline continues running with the remaining samples. It’s also valuable to catch the error and save the problematic row for later inspection. However, this requirement clashed somewhat with API principle #1: Pandas in – Pandas out.
As a solution, error handling is implemented by writing errors to a column named __error__. If you happen to be processing a dataframe that already has a column named __error__, you’ll need to rename it using the RenameColumns link before any error-handling links are applied.
Let’s see this in action with a “toxic” SMILES string:
df_error = pd.DataFrame({"Smiles": ["CCO", "CCC", "☠"]})
df_out = chain(df_error)
df_out
| HeavyAtomCount | ROMol | Smiles | _error_ | |
|---|---|---|---|---|
| 0 | 3.0 | <rdkit.Chem.rdchem.Mol object at 0x7247f1c60970> | CCO | NaN |
| 1 | 3.0 | <rdkit.Chem.rdchem.Mol object at 0x7247f1c60ac0> | CCC | NaN |
| 2 | NaN | NaN | ☠ | Traceback (most recent call last):\n File “/h… |
As it turns out, the smiling skull isn’t a proper chemical SMILES after all. I also found out, It’s even more toxic for WordPress handling of Python code! However, the error is caught and saved in the dataframe, where we can examine the stack trace or error message.
The next logical step is to have a link to filter away or save the rows that failed. We can quickly demonstrate how the StripErrors link works:
from pdchemchain.links import StripErrors error_filter = StripErrors(filename="error_smiles.csv") chain_w_error_filter = chain2 + error_filter df_out = chain_w_error_filter(df_error) df_out
| HeavyAtomCount | ROMol | Smiles | |
|---|---|---|---|
| 0 | 3.0 | <rdkit.Chem.rdchem.Mol object at 0x724828e17bc0> | CCO |
| 1 | 3.0 | <rdkit.Chem.rdchem.Mol object at 0x7247f1cd8270> | CCC |
!cat error_smiles.csv
,HeavyAtomCount,ROMol,Smiles,_error_
2,,,☠,"Traceback (most recent call last):
File ""/home/esben/git/pdchemchain/pdchemchain/base.py"", line 299, in _safe_row_apply
row = self._row_apply(row)
File ""/home/esben/git/pdchemchain/pdchemchain/links/chemistry.py"", line 167, in _row_apply
raise ValueError(f""RDKit Error: {e.errors}"")
ValueError: RDKit Error: [10:56:05] SMILES Parse Error: syntax error while parsing:
[10:56:05] SMILES Parse Error: Failed parsing SMILES '☠' for input: '☠'
"
error_filter.error_df
| HeavyAtomCount | ROMol | Smiles | _error_ | |
|---|---|---|---|---|
| 2 | NaN | NaN | ☠ | Traceback (most recent call last):\n File “/h… |
The error_filter also maintains a copy of the error dataframe, accessible as error_filter.error_df. This is overwritten each time the link is applied, but it can be handy for quick inspection in a Jupyter notebook or other interactive session.
Even More Features: Advanced routing and compound links
Earlier, I mentioned how the pipeline is primarily linear in its construction, which imposes certain restrictions on possible pipeline configurations. However, I did introduce an additional link that allows for more complex branching.
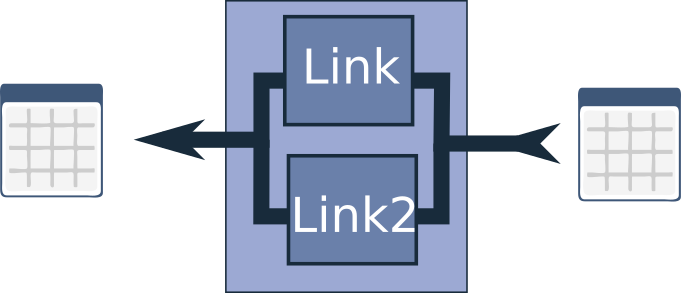
This link is called the UnionLink, inspired in part by the scikit-learn FeatureUnion class. The UnionLink processes the dataframe in two different internal chains and then joins the results back together.
For example, you might want to perform a computationally intensive calculation for only a fraction of your samples. In this case, you can create a chain that filters the dataframe based on a specific value or parameter (hint: the Query link is particularly useful for this), and then add the link with the heavy calculation afterwards. This can serve as one branch in the UnionLink.
The other branch could involve a different kind of processing, or if no additional processing is needed and you simply want to join the original dataframe with the processed subset, you can use the NullLink as the other branch. The two dataframes are then joined together before being returned from the UnionLink.
It’s worth noting that there’s no limitation on nesting UnionLinks within each other, and you can even add other links or chains between them. This flexibility stems from the fact that a chain is a link, and a UnionLink is also a link.
But there’s more to consider. While the UnionLink doesn’t run its two internal links in parallel or on different processors (they run sequentially), the idea of parallel processing isn’t far off the mark. In fact, there is a link designed for running chunks of the dataframe in parallel across different processes: the ParallelPartitionProcessor.
Another issue I’ve encountered is high memory usage. Pandas is inherently memory-hungry, which can be problematic when working with large dataframes. To address this, I’ve implemented the SerialPartitionProcessor. This link also chunks up the dataframe into smaller portions before applying the link or chain, but it processes these chunks sequentially, which can significantly reduce memory consumption.
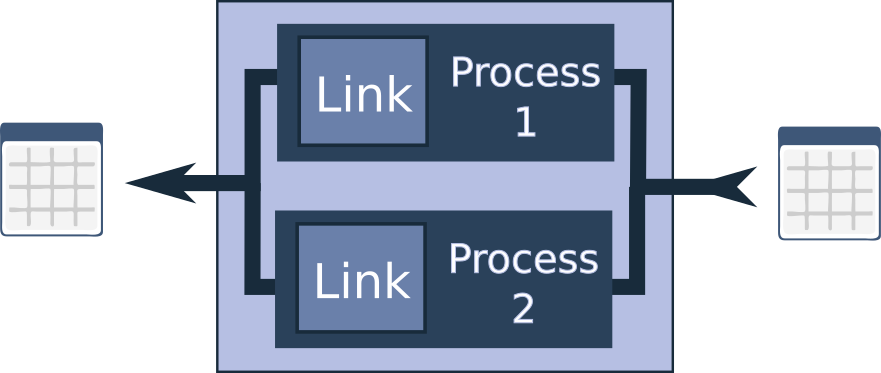
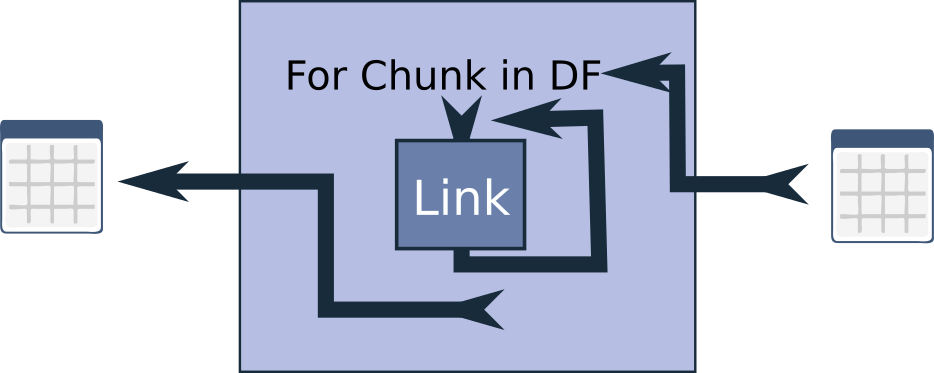
A particularly effective strategy is to nest a ParallelPartitionProcessor inside a SerialPartitionProcessor. This approach allows you to save memory while still accelerating processing by handling smaller chunks in parallel. The results are then joined back together. It’s worth noting that there is some overhead involved in distributing the work and collecting the results, so this approach may not be beneficial for smaller dataframes or very small chunk sizes. However, for larger dataframes, it can be a powerful tool.
Additionally, I’ve included DropColumns and KeepColumns links that can be used to streamline the dataframe before it’s returned from parallel processes and undergoes final concatenation. For instance, if you no longer need the ROMol column, dropping it before the final join can further reduce memory usage and speed up the concatenation process.
By employing these techniques, the pipeline becomes capable of processing millions of rows on a single workstation with a reasonable amount of RAM. This scalability significantly enhances the utility of the package for large-scale data processing tasks in chemical informatics.
Custom Links
But what if I didn’t anticipate the specific Link you need for your unique use case? No problem – you can easily create your own links.
Personally, I have a strong aversion to excessive boilerplate code. So, I put considerable effort into minimizing the overhead required to write new links. I believe the result is close to the minimal possible complexity.
Let me quickly demonstrate how to write a new link that multiplies a column by 2. (It’s admittedly a simplistic example, but it’s easy to comprehend. For these basic arithmetic operations, it would actually be more efficient to use the DfEval or DfRowEval links).
from pdchemchain.links import RowLink
from dataclasses import dataclass
@dataclass
class MySpecialAndPreciousLink(RowLink):
in_column: str
out_column: str
def _row_apply(self, row: pd.Series) -> pd.Series:
row[self.out_column] = row[self.in_column] * 2
return row
link = MySpecialAndPreciousLink(in_column="HeavyAtomCount", out_column="HeavyAtomCount_x2")
df_out = link(df_out)
df_out
| HeavyAtomCount | ROMol | Smiles | HeavyAtomCount_x2 | |
|---|---|---|---|---|
| 0 | 3.0 | <rdkit.Chem.rdchem.Mol object at 0x724828e17bc0> | CCO | 6.0 |
| 1 | 3.0 | <rdkit.Chem.rdchem.Mol object at 0x7247f1cd8270> | CCC | 6.0 |
This example illustrates the straightforward process of creating custom links. However, a full walkthrough on customizations would likely be better suited for a separate blog post, as there’s quite a bit more to explore in that area.
This example illustrates the straightforward process of creating custom links. Let’s break down the additional code required:
- One line for the dataclass decorator
- One line for the class definition
- Two (or more) lines for the parameters
- One line for overloading the _row_apply() method
- One line to return the row
In total, that’s just 6 lines of extra code. For this minimal overhead, you get a fully functional, self-documenting, and auto-configurable link with built-in error handling. This link can be seamlessly integrated into complex pipelines and parallelized if needed.
I have some thoughts on the implementation:
The name ‘dataclass’ for the class decorator is somewhat misleading, as its utility extends far beyond just dataclasses. As example, the parameter definition method is particularly convenient, and the way parameters are stored in fields proved very useful when implementing the auto-documenting and self-configurability features.
The RowLink class is designed for links that process dataframes row by row. While this approach does introduce some overhead, it’s should be the preferable choice to subclass for chemical conversions and calculations, which tend to be more computationally intensive than simple numeric or string operations. The ease of working with rows and adding new columns using dictionary-like assignments is a significant advantage. Moreover, the RowLink class comes with built-in error handling – you can simply raise an exception with your error message (and unexpected exceptions are also handled ;-).
The alternative is to directly subclass the Link abstract class, which provides full access to the dataframe in the abstract apply() method to override. However, this approach requires substantially more code to achieve the same result, as errors must also be handled explicitly in the abstract method. Despite some convenience functions for working with errors and row log messages (.append_errors and .append_log), I’ve found that the amount of code needed to achieve the same functionality is much larger than with the RowLink class.
For completeness, if you wanted to use the DfEval or DfRowEval links for the same purpose, you would do it like this:
from pdchemchain.links import DfEval, RowEval link = DfEval(out_column="HeavyAtomCount_x2_eval", eval_str="HeavyAtomCount * 2") link2 = RowEval(out_column="HeavyAtomCount_x2_roweval", eval_str="row.HeavyAtomCount * 2") df_out = (link + link2)(df_out) df_out
| HeavyAtomCount | ROMol | Smiles | HeavyAtomCount_x2 | HeavyAtomCount_x2_eval | HeavyAtomCount_x2_roweval | |
|---|---|---|---|---|---|---|
| 0 | 3.0 | <rdkit.Chem.rdchem.Mol object at 0x724828e17bc0> | CCO | 6.0 | 6.0 | 6.0 |
| 1 | 3.0 | <rdkit.Chem.rdchem.Mol object at 0x7247f1cd8270> | CCC | 6.0 | 6.0 | 6.0 |
Note the slightly different syntax for the eval_str in these cases.
Final thoughts
I hope I’ve given some overall feel for the thoughts, principles, capabilities and approach behind the pdchemchain package. I think there’s both pros and cons to the design, but overall I find it pretty useful for my own work. It may not be the right tool for all tasks, but as I mentioned in the start of the post, there are many other pipelining tools out there. To summarize for pdchemchain:
Pros
- Simple and fast building for interactive usage in e.g. Jupyter notebooks and IPython settings. Ideal for exploratory data analysis.
- Configurable command line usage via JSON or Yaml files for server side or cluster node usage via the command-line interface
- Interactive and configurable are fully interchangable
- Easy to extend with new Link classes, minimal overhead when writing new links.
Cons
- The simplistic pipeline creation and dogmatic API gives some restrictions (e.g. for outputting both a dataframe and a dataframe with errors)
- The framework are aimed for in_memory usage, so very large dataframes can give issues as pandas usually works on copies. However, the SerialPartitionProcesser wrapper link can significantly reduce memory issues, and links like DropColumns or KeepColumns can also be used to reduce dataframe size.
- Most links work row by row, and cross-row calculations may not be fully supported (e.g. if using partitioning links or the UnionLink). Aggregation of columns values or grouping operations will thus probably not be compatible with all other links.
- Certain names for columns are not allowed as they are needed for internal usage, so avoid dunder column names starting and ending with double underscores e.g. __error__
- It may not be able to build all types of fully directed acyclic graph.
All there is left to say is Happy Hacking!, hope to see interesting links and applications coming out of this 🙂
/Esben
Here’s the link for the GitHub Repo https://github.com/EBjerrum/pdchemchain