Teaching Computers Molecular Creativity
Neural Networks are interesting algorithms, but sometimes also a bit spooky. In this blog post I explore the possibilities for teaching the neural networks to generate completely novel drug like molecules.
I have experimented for some time with recurrent neural networks with the LSTM architecture. In short, recurrent neural networks differ from more traditional feed forward neural networks because they do not take a fixed number of features, but rather gets a sequence of the same feature. Thus they have some interesting applications when it comes to analysis of for example time series such as stock quotes or text of non fixed length. The neurons are stacked in deep layers and feed the state of the network continuously forward to the next analyzed feature in the sequence.
The LSTM (Long Short Term Memory) is a micro architecture of the neurons, which are pre designed to better help with remembering previous states. By priming the neural network with a the special connections the recurrent network more easily learn and can better use information from longer back in the sequence. All this is much better explained in this excellent blog post by Andrej Karpathy, where the text generating properties of the networks are explored (Shakespeare and Linux kernel source code ;-).
After training the recurrent neural networks it is possible to get it to generate novel text. After feeding it a starter sequence, it will output a probabilities for the next characters in the sequence. This can be samples by selecting one from the most probably and then feeding this back to the network, predict the next, feed it back to the network and so forth. This generates novel text!
In chemoinformatics we have the SMILES representation of molecules. SMILES is an acronym for Simplified molecular-input line-entry system, details can be found on the Wikipedia page on the subject. In short its a way to specify a molecule as a single line of text.
So the idea was simply that (Smiles = molecules as text) + Recurrent Neural Networks + Database of drug like molecules => Computational Molecular Creativity!
The Char-RNN code written by Andrej Karpathy was downloaded and installed on my Linux workstation for initial testing. However I ended up using torch-rnn. As molecules I chose a subset of the ZINC database. A simple RDKit script was used to convert the first 600.000 molecules to canonical smiles. After tuning the learning rate, the algorithm was able to output text samples such as this:
th sample.lua -temperature 0.9 -gpu -1 -length 2000 -checkpoint cv/checkpoint_74000.t7 4C=CC=C4)S1)CC1=CSC=C1 CC1=CC(NC(=O)COC2=CC=C3C(=C2)N=NN3C2=CC=C(Cl)C=C2)=CC=C1F CC1=CC=C(C(=O)N2CCN(C(=O)CCN3C(=O)N(C)C4=NC5=C(C=CC=C5)C4=O)CCC3=CC=CC=C32)C=C1 CC(C(=O)NCC1=CC=CC(N2C=NN=N2)=C1)N1C=CN=C1 CC(NC(=O)NCC(C)(C)C1=NC2=C(C=CC=C2)S1)C1=CC=CC=C1
It seems like it have difficulties generating the first line, which is probably because I gave it no starter sequence. However, the neural network quickly gets up to speed and starts to generate novel smiles strings. Another RDKit script let me visualize the molecules.
#Call the Network and show the 16 first sanitizable molecules
import sys
from rdkit import Chem
from rdkit.Chem import Draw
if len(sys.argv) == 2:
fname = sys.argv[1]
else:
print "Need to specify checkpoint t7 file"
sys.exit()
command = 'th sample.lua -temperature 1 -gpu -1 -length 2000 -checkpoint %s'%fname
from subprocess import check_output
smiles = check_output(command.split(' ')).split('\n')
print smiles
mols = []
for smile in smiles[1:-2]:
try:
mol = Chem.MolFromSmiles(smile)#, sanitize=False)
mol.UpdatePropertyCache()
if mol != None:
mols.append(mol)
except:
print "Error parsing smile"
conversion = float(len(mols))/(len(smiles)-3)*100
print "Converted %0.2f %% of the smiles"%conversion
img=Draw.MolsToGridImage(mols[:16],molsPerRow=4,subImgSize=(200,200))
img.save('/home/esben/Mol_examples.png')
img.show()
python Show_mols.py cv/checkpoint_74000.t7 [15:37:47] SMILES Parse Error: unclosed ring for input: NC(=O)C1CCN(C2=CC(=O)N3C4=C(C=CC=C3)N=C4C=C3)CCN1C=C2 Error parsing smile [15:37:47] SMILES Parse Error: unclosed ring for input: CC1=CC(OC2=CC(C)=CC(C(=O)OCC(=O)N3C4=CC=CC(C)=C4CC4C)=CC2=O)=C(Cl)C=C1 Error parsing smile Converted 94.87 % of the smiles

Molecules generated with a recurrent neural network
Some of them seem a bit funky, but others seem just fine. It could be interesting to hear the opinion from a medicinal chemist. Changing the “temperature” makes the network be more wild and creative in its generation but affects the percentage of sanitizable molecules.
I did a few substructure searches on the generated molecules, and they were not represented in the training data set. However, one of the tested structures was found in the remainder of the data downloaded from Zinc, which support the claim that at least some of the molecules are not just correctly formated smiles but can indeed be synthesized.
But how could this be useful?
Expanding the Chemical Space
Chemical space is a conceptual term to imagine molecules spread out in a multidimensional space defined by their properties, such as lipofilicity, target affinity or what other feature are of interest. The problem is that is there are insanely many molecules that are possible. The number of organic molecules of drug like size with “normal” atoms and up to 4 rings are estimated to be around 1060. https://en.wikipedia.org/wiki/Chemical_space
This is indeed a VERY LARGE NUMBER which arises due to the fantastic possibilities for combinatorics in organic chemistry. In comparison, the estimated number of protons in the observable universe is estimated to be 1080 (Eddington Number) and the number of stars in the universe is estimated to be 1024. So it seems like there are more imaginable drug like molecules than protons in a star: 1056. Our current available databases for in silico screening purposes are ridiculously small in comparison. The Zinc database contains 35 million compounds, which is the equivalent of approximately 10-15 grams of hydrogen atoms. Note the minus sign, its not even a nanogram (10-9), in fact its 0.000001 nanograms.
So next time somebody says they screened a large database of compounds you are allowed to laugh out loud ;-).
So getting neural networks to generate new molecules could possible be used to expand the space covered by known compounds. However, it is of little use if it just generates molecules all over the place, so I went further and tried to get the neural network to focus on an area of chemical space.
Learning Transfer in Molecular Creativity
I had an old dataset with DHFR inhibitors lying around. The dataset was probably to small to be used to train the neural network on its own, but by modifying the source code for the neural network to fix the internal dictionary, it was possible to first train the neural network on the Zinc database, and then retrain the network on the much smaller dataset with DHFR inhibitors, the performance was much better in the later case.
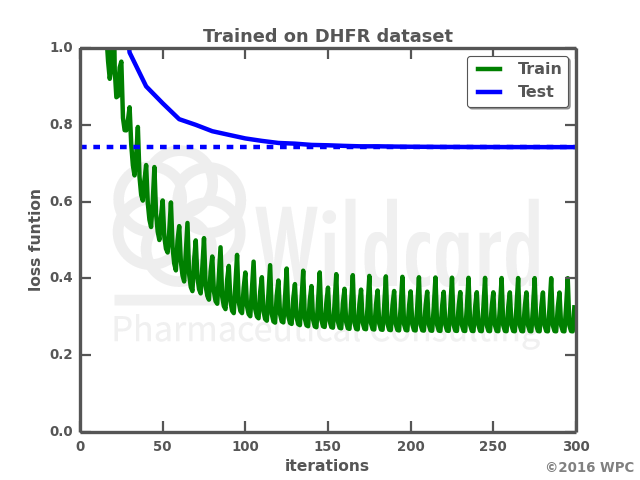
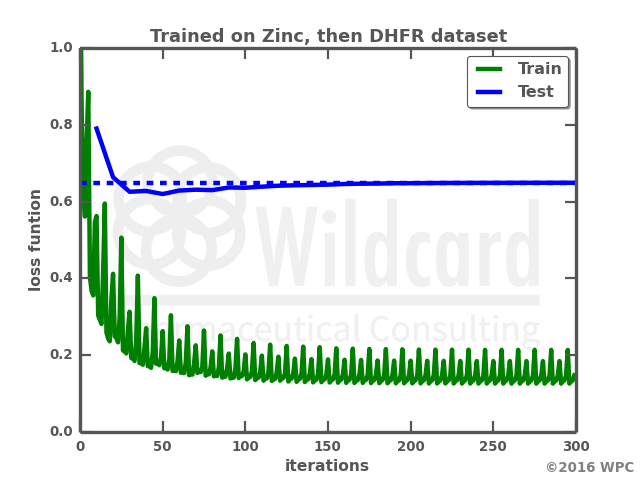
The graphs show the performance over time when training the network on the DHFR dataset on its own (left), or retraining a previously trained network. Not only does the retraining result in a much faster training, the loss function also gets lower on both the train data and the test data. I actually anticipated that training on the small dataset would result in a total overfit of the neural network with a loss function of zero. However, the network trained only on the small dataset actually struggles with generating valid smiles, where I would have expected it just to “memorize” the train data smiles. Below is a comparison of the molecules output by the two networks. 
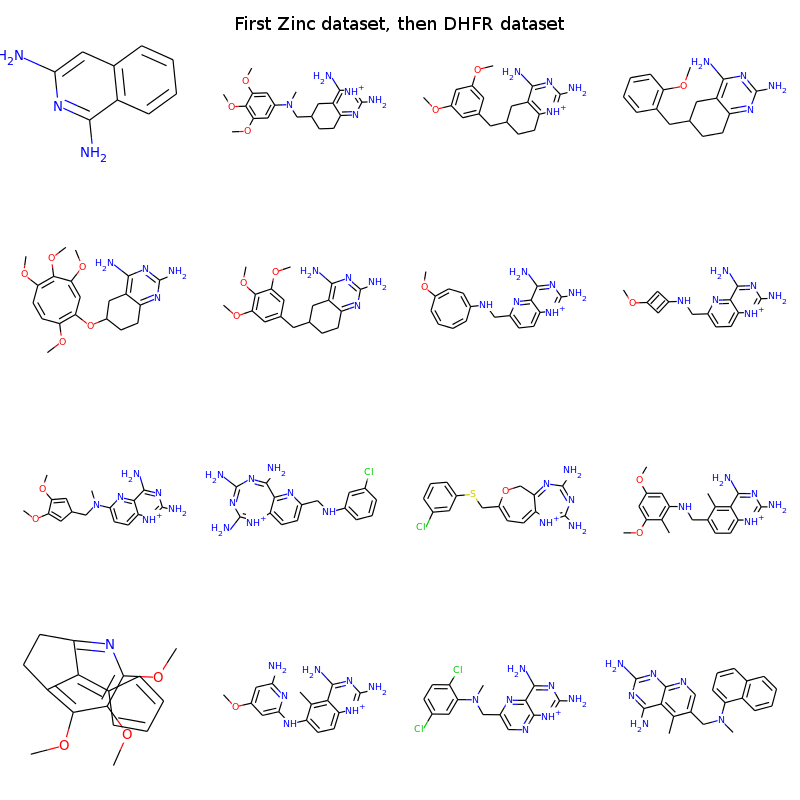 The first one outputs rather small molecules and has a low rate of valid smiles. Most smiles that are not valid, are due to parentheses with no counterpart and numbers with no partner (failed ring closures). The shorter smiles have a higher chance of NOT having these problems. However the retrained neural network clearly outputs molecules which look a bit like DHFR inhibitors. If you know the SAR of the DHFR inhibitors it is possible to see examples of the warhead, the linker and end groups. Some of them look a bit off, whereas others may be plausible candidates? With a bit more tuning the neural networks could probably be nice molecular generators for molecules lying in the vicinity of known compounds of interest.
The first one outputs rather small molecules and has a low rate of valid smiles. Most smiles that are not valid, are due to parentheses with no counterpart and numbers with no partner (failed ring closures). The shorter smiles have a higher chance of NOT having these problems. However the retrained neural network clearly outputs molecules which look a bit like DHFR inhibitors. If you know the SAR of the DHFR inhibitors it is possible to see examples of the warhead, the linker and end groups. Some of them look a bit off, whereas others may be plausible candidates? With a bit more tuning the neural networks could probably be nice molecular generators for molecules lying in the vicinity of known compounds of interest.
Further Ideas
It could be interesting to couple these new molecular generators to in silico screening methods such as docking or QSAR models. The high scoring compounds could then be fed back to the dataset used for re-training and focusing the neural network. I’m interested in starting up research collaborations to investigate this, so please take contact if you are interested collaborating and explore these interesting networks further 😉
It could also be interesting to test if it could be used as a Patent Busting algorithm:
- Retrain Neural Network on Patent data
- Generate molecules
- Filter away molecules covered by patents
- …
- Profit
I’m quite impressed by the ease and speed these neural networks could be trained. However, I’m slightly worried that the generated molecules are not synthetically feasible or posses unwanted properties. So please comment if you know anything about such matters 😉
While writing this Blog post I became aware that this is also the topic of a article from technology review, it certainly seem to be a hot topic with deep neural networks as drug designers. https://www.technologyreview.com/s/602756/software-dreams-up-new-molecules-in-quest-for-wonder-drugs/
UPDATE: 2017-05-24: Check out the preprint I wrote regarding the molecular properties of the produced molecules (using model built with Keras): https://arxiv.org/abs/1705.04612 I’ll write a blog post soon.
This is an amazing work on how machine learning techniques can be successfully applied over some topics that require some revolution, as computational chemistry or in this particular case, what peers in the field call chemo-informatics.
Taking a look to the generated molecules, many of them, for me, exhibit unsuitable properties, but it’s extremely easy to compute many of the desired properties from a computational viewpoint and in a minor fraction of time.
As you pointed out, the big concern here is the synthetic feasibility, as many of them seem pretty weird, but you can easily filter them out applying some well-known retro-synthetic rules.
I think you work paves the way for future and really promising work, as having virtual access to new chemical spaces is a really good alternative to circumvent many of the limitations not solved in drug discovery yet.
In case you want, we can try to make some prospective work to better understand how ML can be applied in a more fine-tuned way to some medicinal chemistry problems
Thanks, but the idea seem to have emerged multiple places at the same time, so I guess the time was ripe. It could be great to start up a collaboration to have a look into the properties of the generated molecules. I’ll write you on the e-mail you provided 🙂
Amazing post!
We used LSTM for antybody humanization!)
We also had the task of generating molecules. We went two ways:
1. used SMIRKS for filtering real structures
2. trained neural network
Thank you for commenting. I guess with smirks you can much better control what molecules are being generated and probably also their synthetic feasibility? This LSTM approach I wrote about here is a bit more wild and will probably need more carefully cleanup and molecular evaluation. How did you do the training of Neural Network afterwards?
Another great post. I will be studying your work here in detail. I had some trouble getting the molecular-autoencoder working from the code on Github (https://github.com/maxhodak/keras-molecules) – I was trying to do interpolation between two compounds a few weeks ago, and I kept getting non-valid SMILES out. Oddly enough, parenthesis were not closed. Will try again after I finish my current project.
Thanks for commenting. The molecular autoencoder is also a very interesting concept, I wrote a bit about my experiments with a bit-starved model here:
I’ve also heard about others having issues with the interpolation. How did you train the model and on what?
Pingback: Non-conditional De Novo molecular Generation with Transformer Encoders | Cheminformania